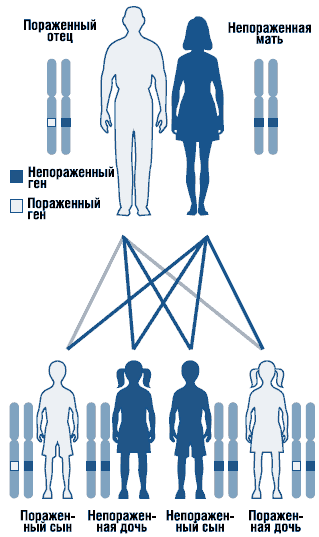
Mutazioni de novo. Rilevazione di una mutazione de novo nel gene della distrofina e suo significato per la consulenza genetica medica nella distrofia muscolare di Duchenne (caso clinico)
Citogenetica medica - lo studio del cariotipo umano in salute e malattia. Questa tendenza è nata nel 1956, quando Thio e Levan hanno migliorato il metodo per preparare i preparati di cromosomi in metafase e per la prima volta hanno stabilito il numero modale di cromosomi (2n = 46) in un set diploide. Nel 1959 fu decifrata l'eziologia cromosomica di una serie di malattie: le sindromi di Down, Klinefelter, Shereshevsky-Turner e alcune altre sindromi da trisomia autosomica. L'ulteriore sviluppo della citogenetica medica alla fine degli anni '60 fu dovuto all'emergere di metodi per la colorazione differenziale dei cromosomi in metafase, che consentivano di identificare i cromosomi e le loro singole regioni. I metodi di colorazione differenziale non sempre garantivano la corretta identificazione dei punti di rottura a seguito di riarrangiamenti strutturali dei cromosomi. Nel 1976, Eunice sviluppò nuovi metodi per studiarli nella fase di prometafase, chiamati "metodi ad alta risoluzione".
L'uso di tali metodi ha permesso di ottenere cromosomi con un diverso numero di segmenti (da 550 a 850) e ha permesso di identificare le violazioni con il coinvolgimento delle loro piccole sezioni (micro-arrangiamenti). Dai primi anni '80. la citogenetica umana è entrata in una nuova fase di sviluppo: l'analisi cromosomica dei metodi citogenetici molecolari, l'ibridazione in situ a fluorescenza (FISH - Fluorescence In Situ Hybridization) è stata introdotta nella pratica. Questo metodo è ampiamente utilizzato per rilevare anomalie cromosomiche strutturali più sottili che sono indistinguibili dalla colorazione differenziale. Attualmente, l'uso di vari metodi di analisi cromosomica consente di eseguire con successo la diagnostica pre e postnatale delle malattie cromosomiche.
Le malattie cromosomiche sono un ampio gruppo di condizioni clinicamente diverse caratterizzate da molteplici malformazioni congenite, la cui eziologia è associata a cambiamenti quantitativi o strutturali nel cariotipo.
Attualmente si distinguono quasi 1000 anomalie cromosomiche, di cui più di 100 forme hanno un quadro clinicamente delineato e sono chiamate sindromi; il loro contributo all'aborto spontaneo, alla mortalità e morbilità neonatale è significativo. La prevalenza di anomalie cromosomiche tra gli aborti spontanei è in media del 50%, tra i neonati con gravi malformazioni congenite multiple - 33%, i nati morti e i decessi perinatali con malformazioni congenite - 29%, neonati prematuri con malformazioni congenite - 17%, neonati con malformazioni congenite - 10%, morti nati morti e perinatali - 7%, bambini prematuri - 2,5%, tutti i neonati - 0,7%.
La maggior parte delle malattie cromosomiche sono sporadiche, che insorgono di nuovo a causa di mutazioni genomiche (cromosomiche) nel gamete di un genitore sano o nelle prime divisioni dello zigote, e non ereditate da generazioni, il che è associato ad un'elevata mortalità dei pazienti nel periodo pre-riproduttivo periodo. La base fenotipica delle malattie cromosomiche è costituita da disturbi dello sviluppo embrionale precoce. Ecco perché i cambiamenti patologici si sviluppano anche nel periodo prenatale dello sviluppo dell'organismo e causano la morte dell'embrione o del feto, o creano il quadro clinico principale della malattia già nel neonato (l'eccezione sono le anomalie dello sviluppo sessuale, che si formano principalmente durante la pubertà). Il danno precoce e multiplo ai sistemi corporei è caratteristico di tutte le forme di malattie cromosomiche. Questi sono dismorfismi craniofacciali, malformazioni congenite di organi interni e parti del corpo, ritardo della crescita e dello sviluppo intrauterino e postnatale, ritardo mentale, difetti del sistema nervoso centrale, cardiovascolare, respiratorio, genito-urinario, digestivo ed endocrino, nonché anomalie ormonali. , stato biochimico e immunologico. Ogni sindrome cromosomica è caratterizzata da un complesso di malformazioni congenite e anomalie dello sviluppo, inerenti in qualche misura solo a questo tipo di patologie cromosomiche. Il polimorfismo clinico di ciascuna malattia cromosomica in forma generale è determinato dal genotipo dell'organismo e dalle condizioni ambientali. Le variazioni nelle manifestazioni della patologia possono essere molto ampie, da un effetto letale a piccole deviazioni nello sviluppo. Nonostante la buona conoscenza delle manifestazioni cliniche e citogenetiche delle malattie cromosomiche, la loro patogenesi, anche in termini generali, non è ancora chiara. Lo schema generale dello sviluppo di processi patologici complessi causati da anomalie cromosomiche e che portano alla comparsa dei fenotipi più complessi delle malattie cromosomiche non è stato sviluppato.
Principali tipi di anomalie cromosomiche
Tutte le malattie cromosomiche per tipo di mutazioni possono essere suddivise in due grandi gruppi: causate da un cambiamento nel numero di cromosomi mantenendo la struttura di quest'ultimo (mutazioni genomiche) e causate da un cambiamento nella struttura del cromosoma (mutazioni cromosomiche ). Le mutazioni genomiche derivano dalla non disgiunzione o dalla perdita di cromosomi durante la gametogenesi o nelle prime fasi dell'embriogenesi. Nell'uomo sono stati trovati solo tre tipi di mutazioni genomiche: tetraploidia, triploidia e aneuploidia. L'incidenza delle mutazioni triploidi (3n = 69) e tetraploidi (4n = 92) è molto bassa, si trovano principalmente tra embrioni o feti abortiti spontaneamente e nei nati morti. L'aspettativa di vita dei neonati con tali disturbi è di diversi giorni. Le mutazioni genomiche sui singoli cromosomi sono numerose e costituiscono la maggior parte delle malattie cromosomiche. Allo stesso tempo, di tutte le varianti di aneuploidia, si verificano solo trisomie da autosomi, polisomie da cromosomi sessuali (tri-, tetra- e pentasomie) e dalle monosomie si verifica solo la monosomia X.
Le trisomie complete o monosomie sono più pesantemente tollerate dall'organismo rispetto a quelle parziali; lo squilibrio nei grandi cromosomi si verifica nei nati vivi molto meno spesso che in quelli piccoli. Le forme complete di anomalie cromosomiche causano anomalie significativamente più gravi di quelle a mosaico. Le monosomie autosomiche tra i nati vivi sono molto rare; sono forme a mosaico con una grande proporzione di cellule normali. Il fatto del valore genetico relativamente basso delle regioni eterocromatiche dei cromosomi è stato dimostrato. Ecco perché si osservano trisomie complete nei nati vivi in quegli autosomi ricchi di eterocromatina - 8, 9, 13, 14, 18, 21, 22 e X. spalla. La monosomia completa sul cromosoma X, compatibile con la vita postnatale, che porta allo sviluppo della sindrome di Shereshevsky-Turner, così come la tetra e la pentasomia, si osserva solo sul cromosoma X, che è eterocromatizzato.
Le mutazioni cromosomiche, o riarrangiamenti cromosomici strutturali, sono disordini del cariotipo, accompagnati o meno da uno squilibrio del materiale genetico all'interno di uno o più cromosomi (riarrangiamenti intra e intercromosomici).
Nella stragrande maggioranza dei casi, le mutazioni cromosomiche strutturali vengono trasferite alla prole da uno dei genitori, nel cui cariotipo c'è un riarrangiamento cromosomico equilibrato. Questi includono la traslocazione bilanciata reciproca (reciproca) senza la perdita di parti dei cromosomi coinvolti in essa. Come l'inversione, non causa fenomeni patologici nell'ospite. Tuttavia, durante la formazione di gameti di portatori di traslocazioni e inversioni bilanciate, si possono formare gameti sbilanciati. Una traslocazione robertsoniana - una traslocazione tra due cromosomi acrocentrici con la perdita dei loro bracci corti - si traduce in un cromosoma metacentrico invece di due acrocentrici. I portatori di tale traslocazione sono sani, perché la perdita dei bracci corti di due cromosomi acrocentrici è compensata dal lavoro degli stessi geni nei restanti 8 cromosomi acrocentrici. Durante la maturazione delle cellule germinali, la distribuzione casuale (durante la divisione cellulare) di due cromosomi riarrangiati e dei loro omologhi porta alla comparsa di diversi tipi di gameti, alcuni dei quali normali, altri contengono una tale combinazione di cromosomi che, al momento della fecondazione, dà uno zigote con un cariotipo riarrangiato equilibrato, e altri ancora danno cromosomicamente durante la fecondazione zigoti sbilanciati.
Con un corredo cromosomico squilibrato (delezioni, duplicazioni, inserzioni), il feto sviluppa gravi patologie cliniche, solitamente sotto forma di un complesso di malformazioni congenite. La mancanza di materiale genetico provoca malformazioni più gravi dell'eccesso.
Le aberrazioni strutturali si verificano molto meno frequentemente de novo. I genitori di un paziente con una malattia cromosomica sono solitamente cariotipicamente normali. La malattia cromosomica in questi casi nasce de novo a seguito della trasmissione da uno dei genitori di una mutazione genomica o cromosomica che si è verificata una volta in uno dei gameti, o tale mutazione si verifica già nello zigote. Ciò non esclude la ricorrenza di anomalie cromosomiche nei bambini di questa famiglia. Ci sono famiglie soggette a ripetuti casi di non disgiunzione cromosomica. Le mutazioni de novo sono quasi tutte trisomie e monosomie conosciute. Il meccanismo principale per il verificarsi di riarrangiamenti strutturali di qualsiasi tipo è una rottura in uno o più cromosomi con successiva riunificazione dei frammenti risultanti.
Indicazioni cliniche per la diagnosi citogenetica
Il metodo di ricerca citogenetico occupa un posto di primo piano tra i metodi di diagnostica di laboratorio nella consulenza medica e genetica e nella diagnostica prenatale. Tuttavia, si dovrebbe aderire rigorosamente all'obiettivo
indicazioni per indirizzare i pazienti allo studio del cariotipo.
Le principali indicazioni per la diagnosi prenatale:
anomalia cromosomica nel figlio precedente in famiglia;
un bambino nato morto con un'anomalia cromosomica;
riarrangiamenti cromosomici, mosaicismo cromosomico o aneuploidia dei cromosomi sessuali nei genitori;
i risultati di uno studio sul siero del sangue nella madre, che indica un aumento del rischio di anomalie cromosomiche nel feto (gruppo a rischio);
età della madre;
anomalie fetali rivelate durante l'esame ecografico;
sospetto di mosaicismo nel feto durante un precedente studio citogenetico;
sospetto di una sindrome con instabilità cromosomica.
Lo studio del cariotipo durante la diagnosi postnatale è raccomandato se il paziente ha:
amenorrea primaria o secondaria o menopausa precoce;
spermogramma anormale - azoospermia o grave oligospermia;
anomalie clinicamente pronunciate nella crescita (crescita bassa, alta) e nelle dimensioni della testa (micro-, macrocefalia);
genitali anormali;
fenotipo anormale o dismorfismi;
malformazioni congenite;
ritardo mentale o disturbi dello sviluppo;
manifestazioni di delezione/microdelezione/sindrome da duplicazione;
Malattia recessiva legata all'X nelle donne;
manifestazioni cliniche delle sindromi da instabilità cromosomica;
durante il monitoraggio dopo il trapianto di midollo osseo.
Gli studi citogenetici dovrebbero essere eseguiti in una coppia sposata:
con anomalie cromosomiche o varianti insolite di cromosomi nel feto, rilevate durante la diagnosi prenatale;
aborti ripetuti (3 o più); natimortalità, morte fetale neonatale, incapacità di esaminare il feto affetto;
il bambino ha un'anomalia cromosomica o una variante cromosomica insolita;
infertilità di eziologia sconosciuta.
L'indicazione per uno studio citogenetico è la presenza dei parenti del paziente:
riarrangiamenti cromosomici;
ritardo mentale, presumibilmente di origine cromosomica;
perdite riproduttive, malformazioni congenite del feto o nati morti di origine sconosciuta.
Indicazioni per lo studio FISH:
sospetto di sindrome da microdelezione, per la quale è disponibile la diagnostica molecolare-citogenetica (presenza di opportune sonde di DNA);
un aumento del rischio di sindrome da microdelezione secondo i dati anamnestici;
segni clinici che suggeriscono mosaicismo per una certa sindrome cromosomica;
condizioni dopo il trapianto di midollo osseo, quando il donatore e il ricevente sono di sesso diverso;
sospetto di un'anomalia cromosomica su uno studio citogenetico standard, quando il metodo FISH può essere utile per ulteriori
chiarimento della natura dell'anomalia, o in situazioni in cui sono presenti manifestazioni cliniche caratteristiche;
la presenza di un cromosoma marker soprannumerario;
sospetto di riarrangiamento cromosomico latente.
Il metodo FISH nell'analisi delle metafasi è mostrato:
con cromosomi marcatori;
materiale aggiuntivo di origine sconosciuta sul cromosoma;
riarrangiamenti cromosomici;
sospetta perdita di un segmento cromosomico;
mosaicismo.
Il metodo FISH nell'analisi dei nuclei interfase è mostrato:
con anomalie cromosomiche numeriche;
duplicazioni;
divisioni;
riarrangiamenti dei cromosomi;
determinazione del sesso cromosomico;
amplificazione genica.
Metodi di ricerca citogenetica:
Lo studio e la descrizione delle caratteristiche dei cromosomi in metafase sono particolarmente importanti per la citogenetica pratica. I singoli cromosomi all'interno di un gruppo vengono riconosciuti utilizzando tecniche di colorazione differenziale. Questi metodi consentono di rilevare l'eterogeneità della struttura cromosomica lungo la lunghezza, determinata dalle peculiarità del complesso dei principali componenti molecolari dei cromosomi: DNA e proteine. Il problema del riconoscimento dei singoli cromosomi in un cariotipo è importante per lo sviluppo della diagnosi citogenetica delle malattie cromosomiche nell'uomo.
I metodi di ricerca citogenetica sono divisi in diretti e indiretti. I metodi diretti vengono utilizzati nei casi in cui è necessario un risultato rapido ed è possibile ottenere preparazioni di cromosomi di cellule che si dividono nel corpo. I metodi indiretti includono, come passaggio obbligatorio, la coltura più o meno prolungata delle cellule in terreni nutritivi artificiali. I metodi che includono la coltivazione a breve termine (da diverse ore a 2-3 giorni) occupano una posizione intermedia.
L'oggetto principale della ricerca citogenetica con metodi diretti e indiretti è lo stadio metafase della mitosi e vari stadi della meiosi. La metafase della mitosi è l'argomento principale della ricerca citogenetica, poiché è in questa fase che è possibile l'identificazione accurata dei cromosomi e l'identificazione delle loro anomalie. I cromosomi nella meiosi vengono esaminati per rilevare alcuni tipi di riarrangiamenti, che per loro natura non si trovano nella metafase della mitosi.
Materiale biologico per studi citogenetici. Elaborazione della coltura cellulare. Preparazione di preparati cromosomici
Le cellule di qualsiasi tessuto disponibile per la biopsia possono essere utilizzate come materiale per ottenere cromosomi umani e studiarli. Molto spesso vengono utilizzati sangue periferico, fibroblasti cutanei, midollo osseo, cellule del liquido amniotico, villi coriali. I più accessibili per lo studio dei cromosomi sono i linfociti del sangue periferico umano.
Attualmente, praticamente in tutti i laboratori del mondo, viene utilizzato un metodo che utilizza sangue intero periferico per la messa in scena di una coltura di linfociti. Il sangue nella quantità di 1-2 ml viene prelevato in anticipo dalla vena cubitale in una provetta sterile o in un flacone con soluzione di eparina. In una fiala, il sangue può essere conservato per 24-48 ore in frigorifero a una temperatura di 4-6 ° C. L'impostazione della coltura dei linfociti viene effettuata in un apposito box room o in una sala di lavoro sotto cappa a flusso laminare in condizioni sterili. Tali condizioni sono obbligatorie per prevenire l'introduzione di flora patogena nell'emocoltura. Se si sospetta una contaminazione di sangue o altro materiale, è necessario aggiungere antibiotici alla miscela di coltura. Le fiale con la miscela di coltura vengono incubate in un termostato a una temperatura di +37 ° C per 72 ore (crescita attiva e divisione cellulare in corso). Lo scopo principale delle tecniche metodologiche nell'elaborazione delle colture cellulari e nella preparazione dei preparati cromosomici da esse è ottenere sulla preparazione un numero sufficiente di piastre metafase con una tale dispersione di cromosomi, alla quale è possibile stimare la lunghezza, forma e altre caratteristiche morfologiche di ciascun cromosoma dell'insieme.
L'accumulo di cellule nella metafase della mitosi e la produzione di piastre di alta qualità sulla preparazione avviene utilizzando una serie di procedure sequenziali:
colchinizzazione - esposizione delle cellule a colchicina o colcemide citostatici, che bloccano la mitosi allo stadio di metafase;
ipotensione delle culture;
fissare le cellule con una miscela di alcol metilico e acido acetico;
applicare una sospensione cellulare su un vetrino.
La colquinizzazione delle colture cellulari viene effettuata 1,5-2 ore prima dell'inizio della fissazione. Dopo la somministrazione di colchicina, le boccette di coltura cellulare continuano ad incubare in un termostato. Al termine dell'incubazione, la miscela di coltura di ciascuna beuta viene versata in provette da centrifuga pulite e centrifugata. Quindi una soluzione ipotonica di cloruro di potassio, preriscaldata a una temperatura di +37 ° C, viene aggiunta al sedimento cellulare.
L'ipotensione viene eseguita in un termostato a una temperatura di +37 ° C per 15 minuti. La soluzione ipotonica di KCI promuove una migliore diffusione dei cromosomi sul vetrino. Dopo l'ipotensione, le cellule vengono sedimentate mediante centrifugazione e fissate. La fissazione viene effettuata con una miscela di alcol metilico (o etilico) con acido acetico.
La fase finale è la preparazione di preparati cromosomici per ottenere piastre metafase ben "diffuse" mantenendo l'integrità, la completezza dell'insieme cromosomico in ciascuna di esse. Una sospensione cellulare viene applicata su vetrini bagnati e raffreddati, dopodiché i bicchieri vengono asciugati a temperatura ambiente e marcati.
Metodi di colorazione dei cromosomi differenziali
Dal 1971, i metodi sono stati ampiamente utilizzati nella citogenetica che consentono di colorare in modo differenziale ciascun cromosoma di un set lungo la sua lunghezza. Il significato pratico di questi metodi risiede nel fatto che la colorazione differenziale consente di identificare tutti i cromosomi umani grazie al modello specifico di colorazione longitudinale per ciascun cromosoma. Qualsiasi colorante costituito da un colorante di base può essere adatto per la colorazione, poiché il principale substrato colorante dei cromosomi è un complesso DNA-proteina. Nella pratica degli studi citogenetici, i seguenti metodi sono i più utilizzati.
Il metodo G-stain è il metodo più comune grazie alla semplicità, affidabilità e disponibilità dei reagenti richiesti. Dopo la colorazione, ogni coppia di cromosomi diventa striata lungo la lunghezza a causa dell'alternanza di segmenti di eterocromatina (scuro) ed eucromatina (chiaro) di colore diverso, comunemente indicati come segmenti G. Il metodo C di colorazione fornisce l'identificazione solo di alcune regioni dei cromosomi. Si tratta di regioni di eterocromatina localizzate nelle regioni pericentromeriche dei bracci lunghi dei cromosomi 1, 9 e 16 e nel braccio lungo del cromosoma Y, nonché nei bracci corti dei cromosomi acrocentrici. Il metodo R di colorazione delle preparazioni cromosomiche mostra un modello di segmentazione differenziale inverso al metodo G. Questo metodo colora bene i segmenti distali dei cromosomi, cosa molto importante per l'identificazione di piccoli riarrangiamenti con coinvolgimento di regioni terminali. Il metodo Q di colorazione fornisce una colorazione fluorescente differenziale dei singoli cromosomi del set, consente di identificare ciascuna coppia di omologhi e anche di determinare la presenza di un cromosoma Y nei nuclei in interfase mediante la luminescenza di un corpo di cromatina Y.
Principi di analisi cromosomica
Una fase obbligatoria dello studio è un'analisi visiva dei cromosomi al microscopio utilizzando un ingrandimento di mille volte (x1000) con oculari x10 e un obiettivo a immersione x100. La valutazione della qualità e dell'idoneità dei preparati cromosomici per la ricerca, nonché la selezione delle piastre metafase per l'analisi viene effettuata a basso ingrandimento (x100). Per la ricerca, scegli piastre metafase complete e ben colorate con una buona dispersione di cromosomi. Il ricercatore calcola il numero totale di cromosomi e valuta la struttura di ciascun cromosoma confrontando la striatura degli omologhi, nonché confrontando il modello osservato con le mappe citogenetiche (schemi) dei cromosomi.
L'uso di sistemi informatici per l'analisi delle immagini facilita notevolmente il compito di un citogenetista, migliora la qualità del suo lavoro e offre l'opportunità di una documentazione rapida e semplice dei risultati della ricerca. Per garantire un'elevata qualità del lavoro, si raccomanda che due specialisti partecipino allo studio citogenetico di ciascun campione. Il documento che conferma lo studio è il protocollo, che indica le coordinate delle cellule scansionate, il numero di cromosomi in ciascuna di esse, i riarrangiamenti rilevati, la formula del cariotipo e la conclusione, nonché il cognome del paziente, la data e il numero di lo studio, il cognome e la firma del medico (medici) che ha condotto lo studio. ... Le diapositive e le immagini dei cromosomi devono essere salvate per una successiva revisione.
REGOLE DI BASE PER LA DESCRIZIONE DELLE ANOMALIE CROMOSOMALI SECONDO IL SISTEMA INTERNAZIONALE DI NOMENCLATURA CITOGENETICA
La registrazione della formula del cariotipo deve essere effettuata secondo la versione attuale del Sistema Internazionale per la Nomenclatura Citogenetica Umana. Di seguito vengono discussi gli aspetti dell'uso della nomenclatura che più spesso si incontrano nella pratica clinica citogenetica.
Il numero e la morfologia dei cromosomi:
In un cariotipo, i cromosomi sono divisi in sette gruppi facilmente distinguibili (A-G) in base alla loro dimensione e posizione del centromero. Gli autosomi sono i cromosomi da 1 a 22, i cromosomi sessuali sono X e Y.
Gruppo A (1-3) - grandi cromosomi metacentrici che possono essere distinti l'uno dall'altro per le dimensioni e la posizione del centromero.
Gruppo B (4-5) - grandi cromosomi submetacentrici.
Gruppo C (6-12, X) - cromosomi metacentrici e submetacentrici di medie dimensioni. Il cromosoma X è uno dei cromosomi più grandi di questo gruppo.
Gruppo D (13-15) - cromosomi acrocentrici di medie dimensioni con satelliti.
Gruppo E (16-18) - cromosomi metacentrici e submetacentrici relativamente piccoli.
Gruppo F (19-20) - piccoli cromosomi metacentrici.
Gruppo G (21-22, Y) - piccoli cromosomi acrocentrici con satelliti. Il cromosoma Y non ha satelliti.
Ogni cromosoma è costituito da una fila continua di strisce che si trovano lungo la lunghezza dei bracci del cromosoma in aree (aree) strettamente limitate. Le regioni cromosomiche sono specifiche di ciascun cromosoma e sono essenziali per la loro identificazione. Le strisce e le regioni sono numerate dal centromero al telomero lungo la lunghezza di ciascun braccio. Le regioni sono sezioni di un cromosoma situate tra due strisce adiacenti. Per designare i bracci corti e lunghi dei cromosomi, vengono utilizzati i seguenti simboli: p - braccio corto e q - braccio lungo. Il centromero (sep) è indicato dal simbolo 10, la parte del centromero adiacente al braccio corto è p10, al braccio lungo - q10. L'area più vicina al centromero è indicata come 1, l'area successiva come 2 e così via.
Per designare i cromosomi, vengono utilizzati simboli a quattro cifre:
1° carattere - numero di cromosomi;
2° carattere (p o q) - braccio cromosomico;
3° carattere - numero del distretto (sito);
Il 4° carattere è il numero della corsia all'interno di quest'area.
Ad esempio, il record 1p31 indica il cromosoma 1, il suo braccio corto, la regione 3, la corsia 1. Se una corsia è suddivisa in sottobande, viene inserito un punto dopo la designazione della corsia, quindi viene scritto il numero di ciascuna sottobanda. Le sottobande, come le strisce, sono numerate nella direzione dal centromero al telomero. Ad esempio, nella banda 1p31 si distinguono tre sottobande: 1p31.1, 1p31.2 e 1p31.3, di cui la sottobanda 1p31.1 è prossimale al centromero e la sottobanda 1p31.3 distale. Se le sottobande sono ulteriormente suddivise in parti, vengono numerate senza punteggiatura. Ad esempio, la sottobanda 1p31.1 è divisa in 1p31.11,1p31.12, ecc.
PRINCIPI GENERALI PER DESCRIVERE IL CARIOTIPO NORMALE E ANOMALE
Nella descrizione del cariotipo, il primo elemento indica il numero totale di cromosomi, inclusi i cromosomi sessuali. Il primo numero è separato dal resto della voce con una virgola, quindi vengono registrati i cromosomi sessuali. Gli autosomi sono designati solo in caso di anomalie.
Un normale cariotipo umano si presenta così:
46, XX - cariotipo femminile normale;
46, XY - cariotipo maschile normale.
Nelle anomalie cromosomiche, vengono registrate per prime le anomalie dei cromosomi sessuali, seguite dalle anomalie autosomiche in ordine di numero crescente e indipendentemente dal tipo di anomalia. Separare ogni anomalia con una virgola. Le designazioni delle lettere sono usate per descrivere i cromosomi strutturalmente riarrangiati. Il cromosoma coinvolto nel riarrangiamento è scritto tra parentesi dopo il simbolo che indica il tipo di riarrangiamento, ad esempio: inv (2), del (4), r (18). Se due o più cromosomi sono coinvolti nel riarrangiamento, viene inserito un punto e virgola (;) tra le designazioni del numero di ciascuno di essi.
I segni (+) o (-) sono posti davanti a un cromosoma per indicare un'anomalia, indicando un cromosoma aggiuntivo o mancante (normale o anormale), ad esempio: + 21, -7, + der (2). Sono anche usati per indicare una diminuzione o un aumento della lunghezza del braccio del cromosoma dopo il simbolo (p o q); a tal fine, i segni di cui sopra possono essere utilizzati solo nel testo, ma non nella descrizione del cariotipo, ad esempio: 4p +, 5q-. Quando si descrivono le dimensioni di segmenti eterocromatici, satelliti e satelliti, il segno (+) (aumento) o (-) (diminuzione) viene posizionato immediatamente dopo la designazione del simbolo corrispondente, ad esempio: 16qh +, 21ps +, 22pstk +. Il segno di moltiplicazione (x) viene utilizzato per descrivere più copie di cromosomi riarrangiati, ma non può essere utilizzato per descrivere più copie di cromosomi normali, ad esempio: 46, XX, del (6) (q13q23) x2. Per indicare interpretazioni alternative delle anomalie, utilizzare il simbolo (оr), ad esempio: 46, XX, del (8) (q21.1) oi (8) (p10).
I cariotipi dei diversi cloni sono separati da una barra (/). Le parentesi quadre sono poste dopo la descrizione del cariotipo per indicare il numero assoluto di cellule in un dato clone. Per indicare il motivo della presenza di cloni diversi, vengono utilizzati i simboli mos (mosaicismo - linee cellulari originate dallo stesso zigote) e chi (chimera - linee cellulari originate da diversi zigoti), che vengono dati prima della descrizione del cariotipo. Quando si elencano i cariotipi, il clone diploide normale è sempre indicato per ultimo, ad esempio: mos47, XY, + 21/46, XY; mos47, XXY / 46, XY.
Se sono presenti più cloni anomali, la registrazione viene effettuata in ordine crescente di dimensione: il primo è quello riscontrato più frequentemente, poi in ordine decrescente. Il più recente è il clone normale, ad esempio: mos45, X/47, XXX/46, XX. Una notazione simile viene utilizzata in un cariotipo con due cloni normali, ad esempio: chi46, XX / 46, XY. Se nel cariotipo sono presenti due cloni anomali, uno dei quali presenta un'anomalia numerica e l'altro un cambiamento strutturale, viene registrato per primo il clone con l'anomalia numerica. Ad esempio: 45, X / 46, X, i (X) (q10).
Quando entrambi i cloni presentano anomalie numeriche, viene registrato per primo il clone avente l'autosoma con numero di serie inferiore, ad esempio: 47, XX, + 8/47, XX, + 21; un clone con anomalie dei cromosomi sessuali viene sempre messo al primo posto, ad esempio: 47, XXX / 47, XX, + 21.
Il fatto che il cariotipo sia aploide o poliploide sarà evidente dal numero di cromosomi e da ulteriori designazioni, ad esempio: 69, XXY. Tutti i cromosomi modificati devono essere etichettati in relazione al livello di ploidia appropriato, ad esempio: 70, XXY, + 21.
L'origine materna o paterna del cromosoma anomalo è indicata dai simboli mat e pat, rispettivamente, dopo l'anomalia descritta, ad esempio: 46, XX, t (5; 6) (q34; q23) mat, inv (14) ( q12q31) pat; 46, XX, t (5; 6) (q34; q23) mat, inv (14) (q12q31) mat. Se è noto che i cromosomi dei genitori sono normali rispetto all'anomalia data, viene considerata come nuova e indicata dal simbolo denovo (dn), ad esempio: 46, XY, t (5; 6) ( q34; q23) mat, inv (14) ( q12q31) dn.
Descrizione delle anomalie cromosomiche numeriche:
Il segno (+) o (-) viene utilizzato per indicare la perdita o l'acquisizione di un cromosoma aggiuntivo quando si descrivono anomalie numeriche.
47, XX, + 21 - cariotipo con trisomia 21.
48, XX, + 13, + 21 - cariotipo con trisomia 13 e trisomia 21.
45, XX, -22 - cariotipo con monosomia 22.
46, XX, + 8, -21 - cariotipo con trisomia 8 e monosomia 21.
Un'eccezione a questa regola sono le anomalie costituzionali dei cromosomi sessuali, che vengono registrate senza utilizzare i segni (+) e (-).
45, X - cariotipo con un cromosoma X (sindrome di Shereshevsky-Turner).
47, XXY - cariotipo con due cromosomi X e un cromosoma Y (sindrome di Klinefelter).
47, XXX - cariotipo con tre cromosomi X.
47, XYY - cariotipo con un cromosoma X e due cromosomi Y.
48, XXXY - cariotipo con tre cromosomi X e un cromosoma Y.
Descrizione delle anomalie cromosomiche strutturali
Nella descrizione dei cambiamenti strutturali vengono utilizzati sistemi di registrazione sia brevi che dettagliati. Quando si utilizza il sistema corto, vengono indicati solo il tipo di riarrangiamento cromosomico e i punti di rottura. Il tipo di anomalia cromosomica, il cromosoma coinvolto nell'anomalia e i punti di rottura sono registrati tra parentesi. Il sistema conciso non consente di descrivere in modo inequivocabile complessi riarrangiamenti cromosomici, che a volte vengono rivelati nell'analisi dei cariotipi tumorali.
Sistema di designazione conciso per modifiche strutturali
Se entrambi i bracci sono coinvolti nel riarrangiamento risultante da due rotture nello stesso cromosoma, il punto di rottura nel braccio corto viene registrato davanti al punto di rottura nel braccio lungo: 46, XX, inv (2) (p21q31). Quando due punti di rottura sono in un braccio del cromosoma, viene indicato per primo il punto di rottura prossimale al centromero: 46, XX, inv (2) (p13p23). Nel caso in cui nel riarrangiamento siano coinvolti due cromosomi, viene indicato per primo o il cromosoma con numero ordinale inferiore o il cromosoma sessuale: 46, XY, t (12; 16) (q13; p11.1); 46, X, t (X; 18) (p11.11; q11.11).
L'eccezione alla regola sono i riarrangiamenti con tre punti di rottura, quando un frammento di un cromosoma viene inserito nella regione di un altro cromosoma. In questo caso, viene registrato per primo il cromosoma ricevente e per ultimo il cromosoma donatore, anche se si tratta di un cromosoma sessuale o di un cromosoma con un numero di serie inferiore: 46, X, ins (5; X) (p14; q21q25); 46, XY, ins (5; 2) (p14; q22q32). Se il riarrangiamento interessa un cromosoma, vengono indicati per primi i punti di rottura nel segmento in cui si è formata l'inserzione. In caso di inserimento diretto, viene registrato prima il punto di rottura del frammento inserito prossimale al centromero e poi il punto di rottura distale. Con l'inserimento invertito, è vero il contrario.
Per designare le traslocazioni in cui sono coinvolti tre diversi cromosomi, indicare in primo luogo il cromosoma sessuale o cromosoma con numero ordinale inferiore, poi il cromosoma che ha ricevuto un frammento dal primo cromosoma, e, infine, il cromosoma che ha dato il frammento a il primo cromosoma. 46, XX, t (9; 22; 17) (q34; q11.2; q22) - un frammento del cromosoma 9, corrispondente alla regione distale di 9q34, trasferito al cromosoma 22, al segmento 22q11.2, un frammento di il cromosoma 22, corrispondente alla regione distale di 22q11 .2 viene trasferito al cromosoma 17, nel segmento 17q22, e un frammento del cromosoma 17, corrispondente alla regione distale di 17q22, viene trasferito al cromosoma 9, nel segmento 9q34.
Sistema di designazione dettagliato per modifiche strutturali. In conformità con il sistema di designazione dettagliato, i riarrangiamenti strutturali dei cromosomi sono determinati dalla composizione delle bande al loro interno. Tutte le designazioni utilizzate nel sistema conciso sono conservate nel sistema dettagliato. Tuttavia, nel sistema dettagliato, viene fornita una descrizione dettagliata della composizione delle bande nei cromosomi riarrangiati con l'uso di simboli aggiuntivi. I due punti (:) indicano un punto di interruzione e i due punti doppi (: :) indicano un'interruzione seguita da una riunione. La freccia (->) indica la direzione di trasferimento dei frammenti cromosomici. Le estremità dei bracci del cromosoma sono indicate dal simbolo ter (terminale), pter o qter indicano rispettivamente l'estremità di un braccio corto o lungo. Il simbolo sep è usato per indicare un centromero.
Tipi di riarrangiamenti cromosomici
Materiale aggiuntivo di origine sconosciuta. Il simbolo add (dal lat. Additio - addizione) viene utilizzato per indicare materiale aggiuntivo di origine sconosciuta, attaccato a una regione o banda cromosomica. Il materiale extra attaccato alla regione terminale causerà un aumento della lunghezza del braccio del cromosoma. Quando si descrivono cromosomi con materiale aggiuntivo di origine sconosciuta in entrambe le braccia, il simbolo der è posto davanti al numero del cromosoma. Se materiale aggiuntivo sconosciuto viene inserito in un braccio di un cromosoma, per la descrizione vengono utilizzati i simboli ins e (?).
Eliminazioni. Il simbolo del viene utilizzato per indicare le eliminazioni terminali (terminali) e interstiziali:
46, XX, del (5) (q13)
46, XX, del (5) (pter-> q13 :)
Il segno (:) significa che la rottura è avvenuta nella banda 5q13, di conseguenza il cromosoma 5 è costituito da un braccio corto e da una parte di braccio lungo racchiuso tra il centromero e il segmento 5q13.
46, XX, del (5) (q13q33)
46, XX, del (5) (pter-> q13 :: q33-> qter)
Il segno (: :) indica la rottura e la riunificazione delle bande 5ql3 e 5q33 del braccio lungo del cromosoma 5. Il segmento cromosomico tra queste bande è cancellato.
I cromosomi derivati o derivati (der) sono cromosomi che sono sorti a seguito di riarrangiamenti che interessano due o più cromosomi, nonché a seguito di più riarrangiamenti all'interno di un cromosoma. Il numero del cromosoma derivato corrisponde al numero del cromosoma intatto, che ha lo stesso centromero del cromosoma derivato:
46, XY, der (9) del (9) (p12) del (9) (q31)
46, XY, der (9) (: p12-> q31 :)
Il cromosoma derivato 9 è il risultato di due delezioni terminali che si verificano nei bracci corti e lunghi, con breakpoint nelle bande 9p12 e 9q31, rispettivamente.
46, XX, der (5) add (5) (p15.1) del (5) (q13)
46, XX, der (5) (? :: p15.1- "q13 :)
Cromosoma derivato 5 con materiale aggiuntivo di origine sconosciuta attaccato alla banda 5p15.1 e una delezione terminale del braccio lungo distale alla banda 5q13.
Cromosomi dicentrici. Il simbolo del dado è usato per descrivere i cromosomi dicentrici. Un cromosoma dicentrico sostituisce uno o due cromosomi normali. Pertanto, non è necessario indicare i cromosomi normali mancanti.
45, XX, dic (13; 13) (q14; q32)
45, XX, dic (13; 13) (13pter-> 13ql4 :: 13q32- "13pter)
La rottura e la riunificazione si sono verificate nelle bande 13ql4 e 13q32 su due cromosomi omologhi 13, con conseguente formazione di un cromosoma dicentrico.
Duplicazioni. Le duplicazioni sono indicate dal simbolo dup; possono essere diritti e invertiti.
46, XX, dup (1) (q22q25)
46, XX, dup (1) (pter-> q25 :: q22-> qter)
Duplicazione diretta di un segmento tra le bande lq22 e lq25.
46, XY, duplicato (1) (q25q22)
46, XY, dup (1) (pter-> q25 :: q25-> q22 :: q25-> qter) o (pter-> q22 :: q25- "q22 :: q22-> qter)
Duplicazione invertita del segmento tra le bande lq22 e lq25. Va notato che solo un sistema dettagliato consente di descrivere la duplicazione invertita.
Inversioni. Il simbolo inv è usato per descrivere le inversioni para- e pericentriche.
46, XX, inv (3) (q21q26.2)
46, XX, inv (3) (pter-> q21 :: q26.2-> q21 :: q26.2-> qter)
Inversione paracentrica, in cui si è verificata la rottura e la riunificazione delle bande 3q21 e 3q26.2 del braccio lungo del cromosoma 3.
46, XY, inv (3) (p13q21)
46, XY, inv (3) (pter- "pl3 :: q21-> p13 :: q21-> qter)
Inversione pericentrica, in cui si è verificata la rottura e la riunificazione tra la striscia 3p13 del braccio corto e la striscia 3q21 del braccio lungo del cromosoma 3. La regione tra queste strisce, compreso il centromero, è invertita di 180 °.
Inserzioni. Il simbolo ins viene utilizzato per indicare l'inserimento diretto o invertito. Un'inserzione è considerata diritta quando l'estremità prossimale della regione di inserzione si trova nella posizione prossimale rispetto alla sua seconda estremità. L'inserimento invertito pone l'estremità prossimale del sito di inserimento in posizione distale. Il tipo di inserimento (diretto o invertito) può essere rappresentato anche rispettivamente da dir e inv.
46, XX, ins (2) (pl3q21q31)
46, XX, ins (2) (pter-> p13 :: q31-> q21 :: pl3- "q21 :: q31-qter)
L'inserimento diretto, ovvero dir ins (2) (p13q21q31), è avvenuto tra i segmenti 2q21 e 2q31 del braccio lungo e il segmento 2p13 del braccio corto del cromosoma 2. La sezione cromosomica del braccio lungo tra i segmenti 2q21 e 2q31 è inserita nel braccio corto nella regione del segmento 2p13. Nella nuova posizione, il segmento 2q21 rimane più vicino al centromero rispetto al segmento 2q31.
46, XY, ins (2) (pl3q31q21)
46, XY, ins (2) (pterH> pl3 :: q21-> q31 :: pl3-> q21 :: q31- "qter)
In questo caso la regione inserita è invertita, cioè inv ins (2) (p13q31q21). Nel riquadro, il segmento 2q21 è più lontano dal centromero rispetto al segmento 2q31. Pertanto, la posizione dei segmenti rispetto al centromero è cambiata.
Isocromosomi. Il simbolo i è usato per descrivere gli isocromosomi, che sono cromosomi costituiti da due bracci identici. I punti di rottura negli isocromosomi sono localizzati nelle regioni centromeriche p10 e q10.
46, XX, io (17) (q10)
46, XX, i (17) (qter- "q10 :: q10 -> qter)
L'isocromosoma lungo il braccio lungo del cromosoma 17 e il punto di rottura sono indicati in 17q10. Il cariotipo ha un cromosoma normale e un cromosoma 17 riarrangiato.
46, X, io (X) (q10)
46, X, i (X) (qter- "q10 :: q10-> qter)
Un cromosoma X normale e un isocromosoma X lungo il braccio lungo.
I siti fragili (fra) possono apparire come normali polimorfismi e possono essere associati a malattie ereditarie o anomalie fenotipiche.
46, X, fra (X) (q27.3)
Una regione fragile nella sottobanda Xq27.3 di uno dei cromosomi X nel cariotipo femminile.
46, Y, fra (X) (q27.3)
Una regione fragile nella sottobanda Xq27.3 del cromosoma X nel cariotipo maschile.
Un cromosoma marcatore (tag) è un cromosoma strutturalmente alterato, nessuna parte del quale può essere identificata. Se viene identificata una qualsiasi parte del cromosoma anomalo, viene descritta come un cromosoma derivato (der). Quando si descrive un cariotipo, un segno (+) è posto prima del simbolo mar.
47, XX, + mar
Un cromosoma marcatore aggiuntivo.
48, X, t (X; 18) (p11.2; q11.2) + 2mar
Due cromosomi marcatori oltre alla traslocazione t (X; 18).
I cromosomi ad anello sono indicati dal simbolo r, possono essere costituiti da uno o più cromosomi.
46, XX, r (7) (p22q36)
46, XX, r (7) (:: p22-> q36: :)
La rottura e la riunificazione si sono verificate nei segmenti 7p22 e 7q36 con la perdita di regioni cromosomiche situate distalmente a questi punti di rottura.
Se il centromero del cromosoma ad anello è sconosciuto, ma sono noti i segmenti dei cromosomi contenuti nell'anello, i cromosomi ad anello sono definiti derivati (der).
46, XX, der (1) r (1; 3) (p36.1q23; q21q27)
46, XX, der (1) (:: lp36.1-> 1q23 :: 3q21-> 3q27: :)
Traslocazioni. Traslocazioni reciproche
Per descrivere le traslocazioni (t), vengono utilizzati gli stessi principi e regole della descrizione di altri riarrangiamenti cromosomici. Per distinguere i cromosomi omologhi, uno degli omologhi può essere sottolineato con un solo trattino basso (_).
46, XY, t (2; 5) (q21; q31)
46, XY, t (2; 5) (2pter2q21 :: 5q31-> 5qter; 5pter 5q31 :: 2q21-> 2qter)
La rottura e la riunione si sono verificate nei segmenti 2q21 e 5q31. I cromosomi scambiano regioni distali a questi segmenti. Il cromosoma con un numero di serie inferiore è indicato per primo.
46, X, t (X; 13) (q27; ql2)
46, X, t (X; 13) (Xpter-> Xq27 :: 13ql2-> 13qter; 13pter-> 3q 12 :: Xq27-> Xqter)
La rottura e la riunione si sono verificate nei segmenti Xq27 e 13q12. I segmenti distali a questi siti sono stati invertiti. Poiché il cromosoma sessuale è coinvolto nella traslocazione, viene registrato per primo. Nota che la notazione corretta è la seguente: 46, X, t (X; 13), non 46, XX, t (X; 13).
46, t (X; Y) (q22; q1, 1.2)
46, t (X; Y) (Xpter-> Xq22 :: Yq11.2-> Yqter; Ypter-> Yq11.2 :: Xq22-> Xqter)
Traslocazione reciproca tra i cromosomi X e Y con break points Xq22 e Yq11. 2.
Le traslocazioni che coinvolgono interi bracci cromosomici possono essere registrate con punti di interruzione nelle regioni centromeriche p10 e q10. Nelle traslocazioni bilanciate, il punto di rottura nel cromosoma sessuale o nel cromosoma con un numero di serie inferiore è designato p10.
46, XY, t (4; 3) (p10; q10)
46, XY, t (1; 3) (lpteMlpl0 :: 3ql0-> 3qter; 3pter-> 3p40 :: 4q40-> 4qter)
Traslocazione reciproca di interi bracci del cromosoma, in cui i bracci corti del cromosoma 1 si uniscono al centromero con i bracci lunghi del cromosoma 3 e i bracci lunghi del cromosoma 1 si uniscono ai bracci corti del cromosoma 3.
In caso di traslocazioni sbilanciate di interi bracci cromosomici, il cromosoma riarrangiato è designato come un derivato (der) e sostituisce due cromosomi normali.
45, XX, der (1; 3) (p10; q10)
45, XX, der (1; 3) (1pt-> 1p10 :: 3q10-> 3qter)
Un cromosoma derivato, costituito da un braccio corto del cromosoma 1 e un braccio lungo del cromosoma 3. I cromosomi mancanti 1 e 3 non sono etichettati in quanto sono stati sostituiti da un cromosoma derivato. Il cariotipo contiene quindi un cromosoma normale 1, un cromosoma normale 3 e un cromosoma derivato der (l; 3).
traslocazioni robertsoniani
Questo è un tipo speciale di traslocazione risultante dalla fusione centrica dei bracci lunghi dei cromosomi acrocentrici 13-15 e 21-22 con la simultanea perdita dei bracci corti di questi cromosomi. I principi per descrivere le traslocazioni sbilanciate che coinvolgono intere spalle sono applicabili anche alla descrizione delle traslocazioni robertsoniani usando il simbolo (der). Il simbolo rob può essere utilizzato anche per descrivere queste traslocazioni, ma non può essere utilizzato per descrivere anomalie acquisite. I punti di rottura dei cromosomi coinvolti nella traslocazione sono indicati nelle regioni q10.
45, XX, der (13; 21) (q10; q10)
45, XX, rubare (13; 21) (q10; q10)
La rottura e la riunione si sono verificate nei segmenti 13q10 e 21q10 delle regioni centromeriche dei cromosomi 13 e 21. Il cromosoma derivato ha sostituito un cromosoma 13 e un cromosoma 21. Non è necessario indicare i cromosomi mancanti. Il cariotipo contiene un normale cromosoma 13, un normale cromosoma 21 e der (13; 21). Lo squilibrio si verifica a causa della perdita dei bracci corti dei cromosomi 13 e 21.
23 marzo 2015Reprogenetics, il più grande laboratorio genetico degli Stati Uniti, in collaborazione con eminenti scienziati cinesi, un certo numero di istituti e centri medici di New York specializzati nel campo della PGD, hanno pubblicato i risultati di nuovi studi, che affermano che si possono trovare mutazioni negli embrioni dopo la fecondazione in vitro (FIV) ...
Per lo studio è sufficiente una piccola (risparmiatrice) biopsia, solo circa 10 cellule embrionali, mentre la maggior parte delle nuove mutazioni (De Novo) che causano una percentuale sproporzionatamente alta di malattie genetiche possono essere rilevate utilizzando la PGD. L'unicità del metodo risiede nello sviluppo di un nuovo processo di screening originale per l'intero genoma esteso.
Nuove mutazioni (De Novo) si verificano solo nelle cellule germinali e negli embrioni dopo la fecondazione. Di norma, queste mutazioni non sono presenti nel sangue dei genitori e anche uno screening completo dei genitori portatori non sarà in grado di rilevarle. La PGD standard non è in grado di rilevare queste mutazioni perché i test non sono sufficientemente sensibili o si concentrano solo su regioni specifiche molto ristrette del genoma.
"Questi risultati rappresentano un passo importante nello sviluppo dello screening dell'intero genoma per trovare gli embrioni più sani nella PGD", afferma Santiago Munné, Ph.D., fondatore e direttore di Reprogenetics e fondatore di Recombine. "Questo nuovo approccio può rilevare quasi tutti i cambiamenti genomici e quindi eliminare la necessità di ulteriori test genetici durante la gravidanza o dopo la nascita, garantendo al contempo che l'embrione più sano sia selezionato per il trasferimento alla futura madre".
È inoltre scientificamente provato che il nuovo metodo riduce il tasso di errore di 100 volte (rispetto ai metodi precedenti).
"È notevole che nuove mutazioni (De Novo) possano essere rilevate con una sensibilità così elevata e tassi di errore estremamente bassi utilizzando un piccolo numero di cellule embrionali", afferma Brock Peters, Ph.D. e scienziato capo dello studio. "Il metodo sviluppato è efficace non solo da un punto di vista medico, ma anche da un punto di vista economico e non vediamo l'ora di continuare la nostra ricerca in questo settore".
Nuove mutazioni possono portare a gravi disturbi cerebrali congeniti come autismo, encefalopatia epilettica, schizofrenia e altri. Poiché queste mutazioni sono uniche per un particolare spermatozoo e ovulo coinvolto nella creazione dell'embrione, l'analisi genetica dei genitori non può rilevarle.
"Fino al cinque percento dei neonati soffre di malattie causate da un difetto genetico", afferma Alan Berkeley, MD, professore, direttore del Dipartimento di Ostetricia e Ginecologia presso il Fertility Center della New York University. "Il nostro approccio è completo e mira a identificare embrioni perfettamente sani. Ciò può alleviare in modo significativo alcuni dei fattori di stress emotivo e fisico della fecondazione in vitro, in particolare per le coppie a rischio di trasmissione di malattie genetiche".
L'articolo è stato tradotto appositamente per il programma IVF School, basato sui materiali
E.V. Tozliyan, pediatra-endocrinologo, genetista, candidato in scienze mediche, unità strutturale separata "Istituto di ricerca clinica di pediatria" N.I. Pirogov, Ministero della Salute della Federazione Russa, Mosca Parole chiave: bambini, sindrome di Noonan, diagnosi.
Parole chiave: bambini, sindrome di Noonan, diagnostica.
L'articolo descrive la sindrome di Noonan (sindrome di Ulrich-Noonan, sindrome di turneroide con cariotipo normale) - una rara patologia congenita, ereditata in modo autosomico dominante, è familiare, ma ci sono anche casi sporadici. La sindrome presuppone la presenza di un fenotipo caratteristico della sindrome di Shereshevsky-Turner nelle femmine e nei maschi con cariotipo normale. Viene presentata un'osservazione clinica. Vengono mostrate le difficoltà della ricerca diagnostica differenziale, l'insufficiente consapevolezza dei clinici su questa sindrome e l'importanza di un approccio interdisciplinare.
Fatti storici
Per la prima volta, O. Kobylinski menzionò l'insolita sindrome nel 1883 (foto 1).
Il più antico caso clinico conosciuto di sindrome di Noonan, descritto nel 1883 da O. Kobylinski
La malattia è stata descritta nel 1963 dal cardiologo americano Jacqueline Noonan, che ha riportato nove pazienti con stenosi della valvola polmonare, bassa statura, ipertelorismo, declino intellettuale moderato, ptosi, criptorchidismo e disturbi scheletrici. Il dottor Noonan, un cardiologo pediatrico dell'Università dell'Iowa, ha notato che i bambini con un raro tipo di difetto cardiaco - stenosi della valvola polmonare - avevano spesso anomalie fisiche tipiche come bassa statura, collo pterigoideo, occhi distanziati e basso impianto orecchie. Ragazzi e ragazze erano ugualmente stupiti. Il dottor John Opitz, un ex studente di Noonan, ha coniato per primo il termine "Sindrome di Noonan" per descrivere la condizione dei bambini che mostravano segni simili a quelli descritti da Noonan. Più tardi, Noonan scrisse l'articolo "Hypertelorism with Turner Phenotype" e nel 1971 il nome "Noonan Syndrome" fu ufficialmente riconosciuto al Cardiovascular Symposium.
Eziologia e patogenesi
La sindrome di Noonan è una malattia autosomica dominante con espressività variabile (Fig. 1). Il gene della sindrome di Noonan si trova sul braccio lungo del cromosoma 12. L'eterogeneità genetica della sindrome non è esclusa. Sono descritte forme sporadiche e familiari della sindrome con una forma di trasmissione autosomica dominante. Nei casi familiari, il gene mutante viene ereditato, di regola, dalla madre, poiché a causa di gravi malformazioni del sistema genito-urinario, gli uomini con questa malattia sono spesso sterili. La maggior parte dei casi riportati sono sporadici, causati da mutazioni de novo.
... Eredità autosomica dominante
Le combinazioni descritte della sindrome di Noonan con la neurofibromatosi di tipo I in diverse famiglie hanno suggerito una possibile connessione tra due loci indipendenti 17q11.2 del cromosoma 17. In alcuni pazienti sono state rilevate microdelezioni al locus 22q11 del cromosoma 22; in questi casi, le manifestazioni cliniche della sindrome di Noonan sono associate all'ipofunzione timica e alla sindrome di DiGeorge. Alcuni autori discutono il coinvolgimento di presunti geni della linfogenesi nella patogenesi della sindrome per la presenza di anomalie facciali e somatiche simili alla sindrome di Turner e un'alta frequenza di patologia del sistema linfatico.
La causa più comune della sindrome di Noonan è una mutazione nel gene PTPN11, che si trova in circa il 50% dei pazienti. La proteina codificata dal gene PTPN11 appartiene a una famiglia di molecole che regolano la risposta delle cellule eucariotiche ai segnali esterni. Il maggior numero di mutazioni nella sindrome di Noonan è localizzato negli esoni 3,7 e 13 del gene PTPN11, che codificano i domini proteici responsabili della transizione della proteina allo stato attivo.
Le possibili idee sulla patogenesi sono rappresentate dai seguenti meccanismi:
La via RAS-MAPK è una via di trasduzione del segnale molto importante attraverso la quale i ligandi extracellulari - alcuni fattori di crescita, citochine e ormoni - stimolano la proliferazione, la differenziazione, la sopravvivenza e il metabolismo cellulare (Fig. 2). Dopo il legame con il ligando, i recettori sulla superficie cellulare vengono fosforilati nei siti della loro regione endoplasmatica. Questo legame coinvolge proteine adattatrici (es. GRB2) che formano un complesso costitutivo con fattori di scambio di nucleotidi guanina (es. SOS) che convertono RAS inattivo legato al PIL nella sua forma attiva legata al GTP. Le proteine RAS attivate attivano quindi la cascata RAF-MEKERK attraverso una serie di reazioni di fosforilazione. Di conseguenza, l'ERK attivato entra nel nucleo per alterare la trascrizione dei geni bersaglio e regolare l'attività dei bersagli endoplasmatici per indurre risposte cellulari adeguate allo stimolo a breve e lungo termine. Tutti i geni coinvolti nella sindrome di Noonan codificano per proteine che sono parte integrante di questo percorso e le mutazioni che causano malattie di solito amplificano il segnale che viaggia attraverso questo percorso.

... Via di segnalazione RAS-MAPK. I segnali di crescita vengono trasmessi dai recettori attivati dal fattore di crescita al nucleo. Le mutazioni in PTPN11, KRAS, SOS1, NRAS e RAF1 sono associate alla sindrome di Noonan, mentre le mutazioni in SHOC2 e CBL sono associate a un fenotipo simile a Noonan
Caratteristiche cliniche della sindrome di Noonan
Il fenotipo dei pazienti con sindrome di Noonan ricorda la sindrome di Turner: collo corto con piega pterigoidea o bassa crescita dei capelli, bassa statura, ipertelorismo delle rime palpebrali (foto 2). Le microanomalie facciali comprendono l'incisione antimongoloide del palato, gli angoli verso il basso del palato, la ptosi, l'epicanto, i padiglioni auricolari bassi, il padiglione auricolare ripiegato, la malocclusione, l'ugola leporino, il palato gotico, la micrognazia e la microgenia. La gabbia toracica tiroidea con capezzoli ipoplasici, ampiamente distanziati, lo sterno sporge nella parte superiore e affonda nella parte inferiore. Circa il 20% dei pazienti ha una patologia scheletrica moderatamente grave. I più comuni sono deformità toracica a imbuto, cifosi, scoliosi; meno spesso - una diminuzione del numero delle vertebre cervicali e della loro fusione, che ricorda le anomalie nella sindrome di Klippel-Feil.

... Fenotipi della sindrome di Noonan
I pazienti con sindrome di Noonan di solito hanno capelli ricci chiari e spessi con una crescita insolita sulla sommità della testa, ci sono spesso macchie dell'età sulla pelle, ipertricosi, degenerazione delle lamine ungueali, anomalie di eruzione e posizionamento dei denti, tendenza a formare cicatrici cheloidi e aumento dell'elasticità della pelle. Un terzo dei pazienti presenta edema linfatico periferico; più spesso, il linfedema delle mani e dei piedi si manifesta nei bambini piccoli. Un sintomo comune è la patologia della vista (miopia, strabismo, esoftalmo moderato, ecc.). Il ritardo della crescita si verifica in circa il 75% dei pazienti, è più pronunciato nei ragazzi ed è generalmente lieve. Il ritardo della crescita si manifesta nei primi anni di vita, meno spesso c'è un leggero deficit di altezza e peso alla nascita. Fin dai primi mesi di vita, c'è una diminuzione dell'appetito. L'età ossea di solito è in ritardo rispetto all'età del passaporto.
Un sintomo caratteristico della sindrome è il criptorchidismo unilaterale o bilaterale, che si verifica nel 70-75% dei pazienti di sesso maschile, nei pazienti adulti c'è azoospermia, oligospermia, alterazioni degenerative dei testicoli. Tuttavia, la pubertà si verifica spontaneamente, a volte con un certo ritardo. Nelle ragazze, c'è spesso un ritardo nella formazione delle mestruazioni, a volte - irregolarità mestruali. La fertilità può essere normale in entrambi i sessi.
Il ritardo mentale viene rilevato in più della metà dei pazienti, di regola è insignificante. Si notano spesso caratteristiche di comportamento, disinibizione, disturbo da deficit di attenzione. Il linguaggio di solito è più sviluppato rispetto ad altre aree intellettuali. Il grado di declino intellettuale non è correlato alla gravità dei disturbi somatici [Marincheva GS, 1988]. In casi isolati vengono descritte malformazioni del sistema nervoso centrale (idrocefalo, ernie spinali), infarti cerebrali tromboembolici, eventualmente associati ad ipoplasia vascolare.
I difetti degli organi interni nella sindrome di Noonan sono abbastanza tipici. Le più tipiche sono le anomalie cardiovascolari: stenosi valvolare dell'arteria polmonare (circa il 60% dei pazienti), cardiomiopatia ipertrofica (20-30%), anomalie strutturali della valvola mitrale, difetti del setto interatriale, tetrade di Fallot; la coartazione dell'aorta è stata descritta solo in pazienti di sesso maschile.
In un terzo dei pazienti si registrano difetti dell'apparato urinario (ipoplasia renale, raddoppio del bacino, idronefrosi, megauretere, ecc.).
Abbastanza spesso, con la sindrome di Noonan, c'è un aumento del sanguinamento, specialmente durante gli interventi chirurgici nella cavità orale e nel rinofaringe. Si riscontrano vari difetti della coagulazione: insufficienza del sistema piastrinico, diminuzione del livello dei fattori di coagulazione, in particolare XI e XII, aumento del tempo di tromboplastina. Ci sono segnalazioni di una combinazione di sindrome di Noonan con leucemia e rabdomiosarcoma, che può indicare un leggero aumento del rischio di malignità in questi pazienti.
La tabella 1 mostra le caratteristiche del fenotipo nella sindrome di Noonan, che cambiano con l'età del paziente. La tabella 2 mostra la correlazione tra fenotipo e genotipo nella sindrome di Noonan.
Tabella 1... Caratteristiche facciali tipiche nei pazienti con sindrome di Noonan per età
| Fronte, viso, capelli | Occhi | Orecchie | Naso | Bocca | Collo | |
| Neonato* | Fronte alta, attaccatura dei capelli bassa nella regione occipitale | Ipertelorismo, fessure oculari inclinate verso il basso, epicanto | – | Radice ribassata corta e larga, punta rivolta verso l'alto | Solco labiale profondamente incassato, picchi alti e larghi del bordo rosso delle labbra, micrognazia | Pelle in eccesso sulla parte posteriore della testa |
| Toracico (2-12 mesi) | Testa grande, fronte alta e sporgente | Ipertelorismo, ptosi o palpebre spesse e cadenti | – | Radice ribassata corta e larga | – | – |
| Bambino (1-12 anni) | Caratteristiche ruvide, muso lungo | – | – | – | – | – |
| Adolescente (12-18 anni) | viso miopatico | – | – | Il ponte è alto e sottile | – | Evidente formazione di pieghe cervicali |
| Adulto (> 18 anni) | I tratti distintivi del viso sono affinati, la pelle appare sottile e trasparente | – | – | Piega nasolabiale sporgente | – | – |
| Tutte le età | – | Iris blu e verdi, sopracciglia a forma di diamante | Orecchie posteriori basse e ruotate con pieghe spesse | – | – | – |
Tavolo 2... Correlazioni tra genotipo e fenotipo nella sindrome di Noonan *
| Il sistema cardiovascolare | Altezza | Sviluppo | Pelle e capelli | Altro | |
| PTPN11 (circa 50%) | Stenosi più pronunciata del tronco polmonare; meno - cardiomiopatia ipertrofica e difetto del setto interatriale | bassa statura; concentrazione di IGF1 inferiore | I pazienti con N308D e N308S hanno una lieve compromissione o un'intelligenza normale | – | Diatesi emorragica più pronunciata e leucemia mielomonocitica giovanile |
| SOS1 (circa 10%) | Difetto del setto interatriale minore | Crescita maggiore | Meno intelligenza ridotta, sviluppo del linguaggio ritardato | Simile alla sindrome cardio-facciale | – |
| RAF1 (circa 10%) | Cardiomiopatia ipertrofica più grave | – | – | Più voglie, lentiggini, macchie di caffè au lait | – |
| KRAS (<2%) | – | – | Ritardo cognitivo più grave | Simile alla sindrome cardio-cutanea-facciale | – |
| NRAS (<1%) | – | – | – | – | – |
Dati di ricerca di laboratorio e funzionale
Non esistono marcatori biochimici specifici per la diagnosi della sindrome di Noonan. In alcuni pazienti si osserva una diminuzione della secrezione notturna spontanea dell'ormone della crescita con una normale risposta ai test di stimolazione farmacologica (clonidina e arginina), una diminuzione del livello di somatomedina-C e una diminuzione della risposta delle somatomedine alla somministrazione di ormone della crescita.
Criteri di diagnosi
La diagnosi di "sindrome di Noonan" viene fatta sulla base dei segni clinici, in alcuni casi la diagnosi è confermata dai risultati della ricerca genetica molecolare. I criteri per diagnosticare la sindrome includono la presenza di un volto caratteristico (con un cariotipo normale) in combinazione con uno dei seguenti: patologia cardiaca, bassa statura o criptorchidismo (nei ragazzi), pubertà ritardata (nelle ragazze). Per identificare la patologia cardiovascolare, è necessario condurre un esame ecografico del cuore con determinazione dinamica delle dimensioni delle cavità e delle pareti dei ventricoli. La diagnosi prenatale della malattia è possibile con l'aiuto del monitoraggio ecografico, che consente di rilevare difetti cardiaci e anomalie nella struttura del collo.
Diagnosi differenziale
Nelle ragazze, la diagnosi differenziale viene effettuata principalmente con la sindrome di Turner; per chiarire la diagnosi, consente uno studio citogenetico. I segni fenotipici della sindrome di Noonan si trovano in una serie di altre malattie: sindrome di Williams, sindrome di LEOPARD, Dubovitsa, sindrome della pelle cardiofacciale, Cornelia de Lange, Cohen, Rubinstein-Teibi, ecc. Materiale clinico, che è attualmente in fase di sviluppo attivo.
Trattamento
Il trattamento dei pazienti con sindrome di Noonan ha lo scopo di eliminare i difetti del sistema cardiovascolare, normalizzare le funzioni mentali, stimolare la crescita e lo sviluppo sessuale. Per il trattamento di pazienti con displasia delle valvole dell'arteria polmonare, tra gli altri metodi, viene utilizzata con successo la valvuloplastica con palloncino. Al fine di stimolare lo sviluppo mentale, vengono utilizzati agenti nootropici e vascolari. I farmaci volti a stimolare lo sviluppo sessuale vengono mostrati principalmente a pazienti con criptorchidismo. I preparati di gonadotropina corionica sono usati in dosaggi legati all'età. In età avanzata - in presenza di ipogonadismo - preparati di testosterone. Negli ultimi anni, forme ricombinanti dell'ormone della crescita umano sono state utilizzate nel trattamento di pazienti con sindrome di Noonan. I dati clinici sono confermati da un aumento del livello di somatomedina-C e della proteina legante specifica durante la terapia. L'altezza finale dei pazienti che ricevono la terapia con ormone della crescita per lungo tempo, in alcuni casi, supera l'altezza media dei membri della famiglia.
Previsione per la vita è determinata dalla gravità della malattia cardiovascolare.
Profilassi la malattia si basa sui dati della consulenza genetica medica.
Consulenza genetica medica
Nella consulenza genetica medica, si dovrebbe procedere dalla modalità di trasmissione autosomica dominante e dall'alto (50%) rischio di ricorrenza della malattia nella famiglia con forme ereditarie. Per identificare la natura del tipo di eredità, è necessario condurre un esame approfondito dei genitori, poiché la sindrome può manifestarsi con sintomi clinici minimi. Attualmente, la diagnosi genetica molecolare della malattia è stata sviluppata e viene migliorata digitando mutazioni nei geni: PTPN11, SOS1, RAF1, KRAS, NRAS, ecc. Sono in fase di sviluppo metodi per la diagnosi prenatale della malattia.
Osservazione clinica
Il ragazzo G., 9 anni (foto 3), è stato osservato nel luogo di residenza da un genetista con diagnosi di patologia cromosomica, sindrome di Williams (fenotipo peculiare, indurimento dei lembi della valvola mitrale, ipercalcemia una volta ogni 3 anni) ? .

... Caratteristiche del fenotipo di un bambino con sindrome di Noonan (scheletro facciale allungato con "guance paffute", collo corto, pieghe pterigoidee sul collo, naso accorciato con narici aperte in avanti, labbra carnose, mento inclinato, incisione antimongoloide delle fessure oculari, malocclusione , macrostomia)
Denunce, contestazioni memoria ridotta, affaticamento, tassi di crescita ridotti.
Storia famigliare : Genitori russi per nazionalità, non consanguinei e privi di rischi professionali, sani. L'altezza del padre è di 192 cm, l'altezza della madre è di 172 cm Non ci sono stati casi di malattia mentale, epilessia o ritardo dello sviluppo nel pedigree.
La vita e la storia medica : un ragazzo dalla 2a gravidanza (1a gravidanza - m / a), che procede con la minaccia di interruzione per tutto, accompagnata da polidramnios. Il primo parto, puntuale, rapido, peso alla nascita - 3400 g, lunghezza - 50 cm Urlò subito, punteggio sulla scala Apgar - 7/9 punti. Alla nascita, il neonatologo ha prestato attenzione al fenotipo insolito del bambino, è stato raccomandato lo studio del cariotipo, il risultato è stato 46, XY (cariotipo maschile normale). Si sospettava ipotiroidismo congenito, è stato effettuato uno studio del profilo tiroideo, il risultato è stato uno stato tiroideo normale. Quindi il bambino è stato seguito da un genetista con una diagnosi presunta di sindrome di Williams. Il primo periodo postnatale era insignificante. Sviluppo motorio per età, le prime parole - entro l'anno, discorso frasario - a 2 anni 3 mesi.
All'età di 8 anni, è stato consultato da un endocrinologo in merito a tassi di crescita ridotti, affaticamento e disturbi della memoria. L'esame radiografico delle mani ha rivelato un moderato ritardo nell'età ossea (CV) rispetto a quella del passaporto (CV corrispondeva a 6 anni). Lo studio del profilo tiroideo ha rivelato un moderato aumento dell'ormone stimolante la tiroide a un livello normale di T4 libero e altri indicatori; Ultrasuoni della tiroide - nessuna patologia. È stata prescritta la terapia ormonale, seguita dall'osservazione dinamica.
Data l'incertezza della diagnosi nel luogo di residenza, il genetista ha inviato il bambino al Centro consultivo e diagnostico regionale di Mosca per i bambini per chiarire la diagnosi.
Dati oggettivi della ricerca:
Altezza - 126 cm, peso - 21 kg.
Lo sviluppo fisico è al di sotto della media, armonioso. Sds di crescita corrisponde a –1 (la norma è –2 + 2). Caratteristiche del fenotipo (foto 3): scheletro facciale allungato con "guance paffute", collo corto, pieghe pterigoidee sul collo, bassa crescita di peli sul collo, naso accorciato con narici aperte in avanti, labbra carnose, mento inclinato, anti-mongoloide fessure oculari, malocclusione, macrostomia, ipertelorismo del capezzolo, asimmetria toracica, sindattilia cutanea incompleta del 2° o 3° dito dei piedi, marcata ipermobilità delle articolazioni interfalangee, unghie fragili e secche. Gli organi interni erano normali. Sviluppo sessuale - Tanner I (che corrisponde al periodo prepuberale).
Dati di ricerca di laboratorio e funzionale:
L'analisi clinica del sangue e delle urine è la norma.
Analisi del sangue biochimica - indicatori entro i limiti normali.
Profilo tiroideo (TSH) - 7,5 µIU / ml (norma - 0,4-4,0), altri indicatori sono normali.
Ormone della crescita (STH) - 7 ng / ml (norma - 7-10), somatomedina-C - 250 ng / ml (norma - 88-360).
Ultrasuoni della tiroide - nessuna patologia.
Ultrasuoni degli organi interni - nessuna particolarità.
ECG - tachicardia sinusale, la posizione normale dell'asse elettrico del cuore.
EchoCG - MVP del 1 ° grado con rigurgito minimo, ispessimento mixomatoso dei lembi della valvola mitrale, un'ulteriore corda nella cavità ventricolare sinistra.
R-grafia della colonna vertebrale - scoliosi del lato destro della colonna vertebrale toracica di 1 ° grado.
Grafico R delle mani con la presa degli avambracci - età ossea 7-8 anni.
Non sono stati registrati modelli EEG di attività epilettica.
MRI del cervello - nessun cambiamento patologico.
Audiogramma - nessuna patologia.
Diagnostica del DNA: studio di genetica molecolare - non sono state trovate delezioni dei loci studiati della regione critica del cromosoma 7; una mutazione Gly434Ary (1230G> A) è stata trovata nell'undicesimo esone del gene SOS1 (analisi del gene PTPN11 - non sono state trovate mutazioni), che è caratteristica della sindrome di Noonan.
Consulenze specialistiche:
Endocrinologo- ipotiroidismo subclinico, compenso farmacologico incompleto.
Oculista- astigmatismo.
Neurologo- distonia vegetativa-vascolare. Reazioni nevrotiche.
Cardiologo- cardiopatia funzionale.
Chirurgo ortopedico- violazione della postura. Deformazione del torace.
Genetista- Sindrome di Noonan.
Tenendo conto del fenotipo del bambino, dei dati sull'anamnesi, dei risultati di ulteriori studi, è stata fatta la diagnosi della sindrome di Noonan, che è stata confermata dal risultato di uno studio di genetica molecolare.
Pertanto, l'osservazione clinica presentata dimostra la complessità della ricerca diagnostica differenziale, la necessità di integrare i segni individuali nel fenotipo generale di una particolare condizione patologica per una diagnosi mirata e tempestiva di alcune forme di malattie ereditarie, l'importanza dei metodi genetici molecolari per chiarire la diagnosi. La diagnosi tempestiva, il chiarimento della genesi di ciascuna sindrome sono particolarmente importanti, in quanto consentono di trovare l'approccio ottimale al trattamento di queste condizioni, la prevenzione di possibili complicanze (fino alla disabilità del bambino); prevenzione della ricorrenza di malattie ereditarie nelle famiglie colpite (consulenza medica e genetica). Ciò impone la necessità per i medici di varie specialità di orientarsi chiaramente nel flusso della patologia ereditaria.
Bibliografia:
- Baird P., Sindrome di De Jong B. Noonan (fenotipo XX e XY Turner) in tre generazioni di una famiglia // J. Pediatr., 1972, vol. 80, pag. 110-114.
- Hasegawa T., Ogata T. et al. Coartazione dell'aorta e ipoplasia renale in un ragazzo con anomalie della superficie Turner / Noonan e un cariotipo 46, XY: un modello clinico per la possibile compromissione di un putativo gene linfogenico per le stimmate somatiche di Turner // Hum. Genet., 1996, vol. 97, pag. 564-567.
- Fedotova TV, Kadnikova V.A. et al. Analisi genetica clinica e molecolare della sindrome di Noonan. Materiali del VI Congresso della Società Russa di Genetisti Medici. Genetica medica, supplemento al n. 5, 2010, p.184.
- Ward K.A., Moss C., McKeown C. La sindrome cardio-facio-cutanea: una manifestazione della sindrome di Noonan? // Fra. J. Dermatol., 1994, vol. 131, pag. 270-274.
- Municchi G., Pasquino A.M. et al. Trattamento con ormone della crescita nella sindrome di Noonan: rapporto di quattro casi che hanno raggiunto l'altezza finale // Horm. Res., 1995, vol. 44, pag. 164-167.
Da BrainstormWiki
Che cos'è la mutazione?
Per cominciare, non esiste un codice genetico "corretto". Quindi, se io ho un gene e tu ne hai un altro, questo non significa ancora che uno di noi sia un mutante.
Tuttavia, ci sono cambiamenti nel genocodice che portano a problemi evidenti e di solito sono rari; si chiamano mutazioni. Cambiamenti che si verificano in più dell'1% della popolazione, è più corretto chiamare non mutazioni, ma polimorfismi.
Le mutazioni possono essere ereditate dai genitori o possono verificarsi solo in un bambino - in questo caso vengono chiamate de novo
Le mutazioni possono verificarsi già durante lo sviluppo dell'organismo, durante la divisione e la differenziazione cellulare - queste sono chiamate mutazioni somatiche; non possono essere ereditati perché “vivono” nelle cellule del corpo (soma), ma non nelle cellule germinali..
Al contrario, le mutazioni che colpiscono le cellule germinali sono ereditate e vengono chiamate mutazioni germinali
Le mutazioni possono essere silenzioso- lo sono, ma non c'è effetto. Ce n'è una stragrande maggioranza: se ricordi, i geni occupano solo meno del 2% di tutto il DNA. Ognuno di noi è portatore di almeno dozzine di tali mutazioni. Se cadono su sezioni insignificanti di DNA, nessuno noterà nulla.
Le mutazioni possono influenzare una "lettera" del codice genetico - o anche un intero enorme pezzo di DNA. Questo frammento può essere molto spesso perso (delezione) o duplicato (duplicazione) - in questo caso, le cellule cambieranno la quantità di proteine sintetizzate per i geni interessati - la loro dose cambierà
Esistono diversi tipi di analisi e test per diversi tipi di mutazioni e ora proveremo a esaminarli.
Dal grande al piccolo
Aneuplodia - un'anomalia nel numero di cromosomi
La più grande anomalia genetica è un cambiamento nel numero di cromosomi. Sebbene non sia l'autismo, vale la pena iniziare con esso 50 anni fa, è stato scoperto che la sindrome di Down è causata da tre 21 cromosomi, invece di due. Questo è chiamato trisomia; e un cambiamento nel numero generale di cromosomi è aneuplodia. Inoltre, ci sono i cromosomi della trisomia 13 (sindrome di Patau) e 18 (sindrome di Edwards), nonché un certo numero di aneuplodie dei cromosomi sessuali (X e Y)
Tutto questo può essere visto in un microscopio ottico convenzionale - l'analisi si chiama "cariotipo". I cromosomi delle cellule in divisione vengono fotografati, ordinati e descritti. L'immagine mostra il cariotipo di una donna con tre 21 cromosomi.
L'effetto delle trisomie è enorme e evidente in molti processi: sia nella struttura del corpo, sia nei processi fisiologici, e persino nelle proteine caratteristiche nel sangue di una madre portatrice (così funzionano i test di screening ora nel villaggio di Downa )
Perché ci sono solo le trisomie 21, 13 e 18 e i cromosomi sessuali? Questo può accadere con qualsiasi cromosoma, ma il resto non sopravvive nemmeno fino allo stadio di gravidanza evidente. Forse la ragione è che i cromosomi 21, 13 e 18 sono alcuni dei geni proteici più poveri (ricordate, la numerazione è lì per dimensione, sono anche piccoli) e l'aumento della loro dose è relativamente tollerabile. Ciò è confermato dalla tabella seguente: nelle prime fasi dello sviluppo, qualsiasi aneuplodia è possibile e mentre ci avviciniamo al parto di successo, solo questi 3.

Copia numero variazione

CNV è anche duplicazione o assenza di una certa parte del DNA, ma non a livello dell'intero cromosoma, ma di diversi milioni di nucleotidi.
Per rilevarli, viene utilizzato un test chiamato array CGH: ibridazione genomica comparativa, anche FISH e altri Nella nostra pratica vengono spesso chiamati cariotipizzazione molecolare, analisi cromosomica microarray, ecc.
Questi sono ancora enormi pezzi di DNA che possono includere dozzine di geni e strutture di controllo. L'effetto è molto evidente, includendo quasi sempre dismorfismi e alterazioni delle capacità cognitive. Sono designati dalla sezione interessata del codice genetico, ad esempio delezione 2q32 - perdita di un sito nella striscia 32 sul braccio lungo del cromosoma 2 (ricorda la parte 1 e gli "indirizzi" delle aree cromosomiche) Molti noti " sindromi nominali”, ad esempio, sindrome di Williams - delezione 7q11.23
Si pensa che tali mutazioni CNV rappresentino dal 3% al 10% dei casi di autismo. Questo è praticamente l'unico tipo di anomalia genetica che è tranquillamente associato ad alcune forme - sindromiche - di autismo.
Ancora una volta, la cosa principale da sapere sulla CNV nell'autismo: funzionano in una piccola frazione di casi, ma il loro effetto è evidente in tanti aspetti, dai dismorfismi all'intelligenza. Quelli. la presenza di difetti nel CNV è quasi sempre riscontrabile sia direttamente sul viso, sia sotto forma di disturbi piuttosto evidenti come ipotensione sistemica e atassia...
Alcuni CNV noti e documentati

Tuttavia, la disponibilità di dati CNV non garantisce l'autismo come diagnosi. La penetranza non è mai al 100%. Un recente studio estone su un gruppo di persone senza alcuna diagnosi ha mostrato che i portatori di CNV 16p11.2 mostrano dismorfismi della testa, obesità, capacità cognitive compromesse - ma non hanno una diagnosi di autismo, contrariamente ai dati nella tabella sopra;
Esiste un database CNV online che causa l'autismo e le sindromi correlate http://projects.tcag.ca/autism/
Autismo Sindromico
La sindrome autistica è chiamata sindrome autistica quando si ritiene che i sintomi autistici siano causati da un disturbo genetico comprensibile - di solito CNV, ma non solo (vedi X Fragile per esempio). Molti scienziati dicono che questo termine non è corretto. Probabilmente è più corretto dire "autismo di una comprensibile eziologia genetica" o qualcosa del genere.
Un'ottima panoramica delle forme più famose di autismo con una "eziologia genetica comprensibile" - ad es. forme sindromiche di autismo possono essere trovate sul blog di Emily Casanova
- Parte 1: Sindromi di Timothy, Smith-Lemli-Opitz, CHARGE, Cornelia de Lange, Lujan-Fryns
Ancora una volta sulla penetranza
Penetranza - La penetranza è un termine che si riferisce alla percentuale di portatori di una mutazione che mostra (in questo caso) l'autismo. Nella revisione del capitolo precedente, si può vedere che la penetranza per le forme sindromiche di autismo è del 60-90 percento.
Una penetranza del 92% per idic (15) è detta "stordente". Sì, è molto. Questo è più che sufficiente per considerare questa variante genetica come la causa dell'autismo - se si trova in un bambino e i sintomi convergono. Ciò suggerisce anche che se questa mutazione viene esclusa nella PGD, il figlio successivo degli stessi genitori non avrà l'autismo.
SNP

Ora veniamo alle più piccole variazioni genetiche, racchiuse in un solo nucleotide, una “lettera”. Ci sono diversi milioni di punti nel genoma umano dove sono comuni variazioni in una “lettera” del codice genetico; tutto intorno è stabile, ma in questi luoghi specifici - per persone diverse in modi diversi. Inoltre, tali variazioni per qualche motivo sono quasi sempre bialleliche, cioè ci sono solo due varianti di "lettere" su quattro, e sono presenti in una percentuale molto ampia della popolazione - spesso il 60% ha una versione e 40 % ne ha un altro. Questa non è una mutazione, ma un polimorfismo, se ricordi la parte 7. (perché un SNP sia ufficialmente riconosciuto come tale, la sua prevalenza deve essere almeno dell'1%)
Cioè, queste non sono "mutazioni", non errori del DNA... Questa è la norma e le sue varianti.
Tali variazioni sono chiamate SNP (leggi "snip"), designate da numeri come rs2320030, e ne sono state trovate e descritte circa 10 milioni.
Studi di associazione a livello di genoma
Sul chip SNP viene eseguita un'analisi che mostra quali varianti SNP (alleli) sono presenti in una determinata persona. Si tratta di una lastra su cui vengono “stampati” campioni del genoma umano attorno a SNPs popolari, su questa piastra viene applicato il DNA del soggetto (spesso un campione di saliva) e poi si guarda dove si è “bloccato” e dove no. La popolare analisi di 23andme è proprio questo e fornisce circa 1 milione di questi SNP. Questo è chiamato "genotipizzazione" ed è abbastanza economico, motivo per cui è spesso usato ora (e spesso non per affari). È economico perché gli SNP sono ben noti ed è conveniente eseguire questa analisi sul flusso, in grandi quantità.
Questa disponibilità ha generato un tipo speciale di ricerca, Genome-Wide Association Studies (GWAS), in cui viene preso un gruppo di portatori di un tratto ereditario e un gruppo di controllo, tutti sono genotipizzati e quindi vedono se c'è qualche SNP che il primo aveva in una variante. , e il secondo - nell'altro.
Il problema qui è nelle statistiche: poiché ci sono milioni di SNP, c'è un'alta probabilità che vedremo una connessione dove non esiste: solo per esempio, su un gruppo di 30 autisti, ci sarà un tale SNP che abbandonare "correttamente" 30 volte da loro. Se una moneta viene lanciata un milione di volte, salirà al limite :) Puoi combattere questo problema usando metodi statistici e combattere con successo. Nel complesso, tuttavia, i risultati GWAS per l'autismo sono replicati in modo disgustoso.
Cosa significa SNP?

L'effetto dell'una o dell'altra opzione SNP è solitamente trascurabile. Non può essere diversamente, dal momento che sono tutti, per definizione, molto diffusi nella popolazione. Tuttavia, ci sono malattie (ad esempio, la fibrosi cistica) per le quali sono decisive (ma di norma non in un SNP: solitamente la presenza di una variante "cattiva" aumenta statisticamente il rischio del disturbo, diciamo, del 2% )
Nessun SNP è stato trovato associato a un grado significativo di autismo. A proposito, quelli che sono collegati almeno in una certa misura - di regola, non si trovano nei geni, ma negli elementi di controllo del DNA - ad es. non nell'esoma.
Pertanto, tutti i test che utilizzano la genotipizzazione SNP e calcolano un "rischio di autismo" significativo sono molto probabilmente ciarlataneria. Ci sono parecchi siti che offrono di scaricare i risultati di 23andme e ottenere ricette "personalizzate" per il trattamento dell'autismo. Il più famoso è "Dr." Yasko, che promuove ricette da "mutazioni" MTHFR. Ciarlataneria. Ancora una volta, queste non sono mutazioni. MTHFR è un gene importante nel metabolismo dei folati ed è probabilmente associato all'autismo, ma non a livello di SNP per cercarlo.
In tutta onestà, va detto che una combinazione di SNP (centinaia di SNP diversi, ognuno dei quali dà un magro contributo, ma nella somma viene digitato qualcosa di significativo) sembra essere in grado di spiegare la schizofrenia (al contrario dell'autismo) - che quasi le stesse persone stanno cercando di identificare ciò che ha fatto Human Genome Project, e uno sforzo quasi altrettanto titanico. Ma lì stiamo parlando esattamente di un centinaio di tagli.
Mutazioni de novo

Le mutazioni "reali" che non vivono in modo permanente in una popolazione, le mutazioni che compaiono in persone specifiche e vengono trasmesse ai loro figli (o si verificano nei bambini) sono chiamate mutazioni de novo. Possono essere come grandi sezioni (CNV), come piccoli inserimenti e delezioni (indel) o come "lettere" separate. Questo post riguarda gli ultimi due.
Per rilevarli, i test su chip economici non sono adatti, il genoma deve essere letto di seguito o, come viene chiamato, il sequenziamento. Puoi leggere l'intero genoma in generale - il sequenziamento dell'intero genoma (costoso) e solo l'esoma, solo la parte che codifica per le proteine - il sequenziamento dell'esoma (relativamente economico, $ 1000 a persona). Di conseguenza, nella pratica clinica, non viene quasi affatto utilizzato.
In linea di principio, ci sono molte di queste mutazioni de novo: ciascuno dei genitori trasmette al bambino almeno 100 di queste, ma solo una piccola parte cade su siti di DNA significativi. Recenti pubblicazioni affidabili (Iossifov e colleghi 2012) stimano il contributo chiave delle mutazioni de novo a circa il 10% dei casi di autismo. Se eri alla conferenza di maggio, c'è stato un discorso di Nagwa Meguid su "Un nuovo gene e una forma unica di autismo" - esattamente un caso del genere, mutazioni de novo (diverse) nel gene BCKDK.
Tuttavia, in questa semplice storia: qui c'è una mutazione, qui ha colpito un gene che è importante per il funzionamento del cervello, qui abbiamo dimostrato che questo gene influenza l'autismo - c'è un grande mistero. Se, a differenza di Ivan Iosifov, per studiare famiglie con non uno, ma due autistici, come hanno fatto quest'anno Yuen e colleghi, si scopre che riescono a prendere diverse mutazioni dagli stessi genitori.
Riassumere e classificare i tipi di "mutazioni"
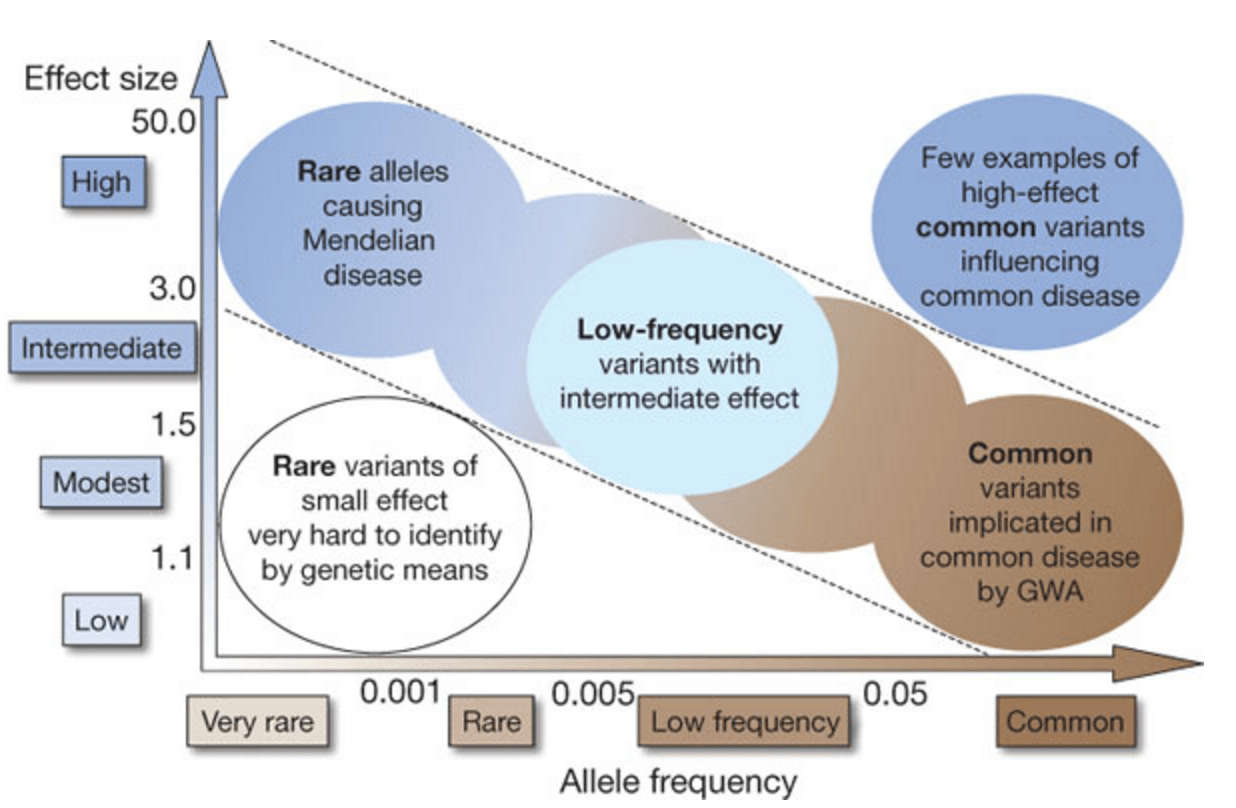
Nelle parti precedenti, abbiamo brevemente esaminato i diversi tipi di varianti genetiche nel DNA. Da grandi dimensioni, solitamente con un effetto notevole, a molto piccole.
E la regola qui è questa: le grandi anomalie sono generalmente rare, ma hanno un effetto notevole su molti aspetti del fenotipo; piccole varianti sono spesso comuni nella popolazione, ma ognuna di esse ha un effetto trascurabile.
Questo può essere riassunto in un diagramma come questo, dove orizzontalmente - la prevalenza dell'allele (variante genetica), da raro a frequente, e verticalmente - l'effetto manifestato nei portatori.
Va notato che per l'autismo e la schizofrenia, anche i CNV più "forti" non hanno un effetto maneliano, aumentando solitamente il rischio di sviluppare una malattia clinicamente determinata dall'1% al 3% -10%. È chiamato penetranza incompleta... Si può essere portatori della ben nota "CNV schizofrenica", si possono avere dismorfismi e anomalie del sistema endocrino caratteristici di essa, ma non avere schizofrenia, sebbene si possano riscontrare alcune anomalie subcliniche.
Di conseguenza, l'effetto individuale dei polimorfismi di tipo SNP è generalmente scarso e non ha quasi alcun significato pratico per la diagnosi di autismo.
| Programma educativo sulla genetica dell'autismo |
|---|
| I. Programma educativo sulla genetica dell'autismo II. Genetica e gemelli III. Tipi di mutazioni IV. |
Tre gruppi di scienziati americani, indipendentemente l'uno dall'altro, sono riusciti per la prima volta a stabilire un legame tra le mutazioni in alcuni geni e la probabilità di sviluppare disturbi dello spettro autistico in un bambino, riporta il New York Times. Inoltre, i ricercatori hanno trovato conferma scientifica di una relazione diretta precedentemente identificata tra l'età dei genitori, in particolare dei padri, e il rischio di sviluppare l'autismo nella prole.
Tutti e tre i gruppi si sono concentrati su un raro gruppo di mutazioni genetiche chiamate "de novo". Queste mutazioni non sono ereditate, ma si verificano durante il concepimento. Come materiale genetico, sono stati prelevati campioni di sangue da membri della famiglia in cui i genitori non erano autistici e i bambini hanno sviluppato vari disturbi dello spettro autistico.
Il primo gruppo di scienziati, guidato da Matthew W. State, professore di genetica e psichiatria infantile alla Yale University, il cui lavoro è stato pubblicato il 4 aprile sulla rivista Nature, ha analizzato la presenza di mutazioni de novo in 200 persone con diagnosi di autismo, i cui genitori , fratelli e sorelle non erano autistici. Di conseguenza, sono stati trovati due bambini con la stessa mutazione nello stesso gene e non erano collegati da nient'altro che dalla diagnosi.
"È come quando si gioca a freccette per colpire lo stesso punto su un bersaglio due volte con un dardo. La probabilità che la mutazione rilevata sia associata all'autismo è del 99,9999 percento", cita il professor State.
Un team guidato da Evan E. Eichler, professore di genetica all'Università di Washington, ha esaminato campioni di sangue di 209 famiglie con bambini autistici e ha trovato la stessa mutazione nello stesso gene in un bambino. Inoltre, sono stati identificati due bambini autistici di famiglie diverse che avevano mutazioni de novo identiche, ma in geni diversi. Non c'erano simili coincidenze tra i soggetti non autistici.
Un terzo gruppo di ricercatori, guidato dal professor Mark J. Daly dell'Università di Harvard, ha trovato diversi casi di mutazioni de novo negli stessi tre geni nei bambini autistici. Almeno una mutazione di questo tipo è presente nel genotipo di qualsiasi persona, ma, secondo Daly, gli autistici, in media, ne hanno molte di più.
Tutti e tre i gruppi di ricercatori hanno anche confermato la relazione precedentemente osservata tra età dei genitori e autismo in un bambino. Più i genitori sono anziani, in particolare il padre, maggiore è il rischio di mutazioni de novo. Dopo aver analizzato 51 mutazioni, il team guidato dal professor Eichler ha scoperto che questo tipo di danno si verifica nel DNA maschile quattro volte più spesso che nel DNA femminile. E ancora più spesso se un uomo ha più di 35 anni. Pertanto, gli scienziati suggeriscono che è il materiale genetico paterno danneggiato ricevuto dalla prole durante il concepimento che è la fonte di quelle mutazioni che comportano lo sviluppo di disturbi autistici.
Gli scienziati concordano sul fatto che la ricerca di modi per prevenire questo sviluppo di eventi sarà lunga, lo studio della natura genetica dell'autismo è solo all'inizio. In particolare, i team di Eichler e Daly hanno trovato prove che i geni in cui si trovano mutazioni de novo sono coinvolti negli stessi processi biologici. "Ma questa è solo la punta dell'iceberg", afferma il professor Eichler. "La cosa principale è che siamo tutti d'accordo su da dove cominciare".










