Cause della malattia. Sintomi e cause della sclerosi laterale amiotrofica
medicina moderna in continua evoluzione. Gli scienziati stanno creando nuovi farmaci per malattie precedentemente incurabili. Tuttavia, oggi gli esperti non possono offrire cure adeguate contro tutti i disturbi. Una di queste patologie è la SLA. Le cause di questa malattia sono ancora inesplorate e il numero di pazienti aumenta solo ogni anno. In questo articolo, daremo un'occhiata più da vicino a questa patologia, ai suoi principali sintomi e ai metodi di trattamento.
Informazione Generale
La sclerosi laterale amiotrofica (SLA, malattia di Charcot) è a patologia grave sistema nervoso, in cui c'è una sconfitta dei cosiddetti motoneuroni nell'area di midollo spinale così come nella corteccia cerebrale. È cronico e malattia incurabile, portando gradualmente alla degenerazione dell'intero sistema nervoso. Sul fasi finali malattia, una persona diventa impotente, ma allo stesso tempo mantiene la lucidità mentale e la salute mentale.
La malattia della SLA, le cui cause e patogenesi non sono state completamente comprese, non è diversa metodi specifici diagnosi e trattamento. Gli scienziati continuano a studiarlo attivamente oggi. Ora è noto con certezza che la malattia si sviluppa principalmente in persone di età compresa tra 50 e circa 70 anni, ma ci sono casi di lesioni precoci.
Classificazione
A seconda della localizzazione delle manifestazioni primarie della malattia, gli esperti distinguono le seguenti forme:
- Forma lombosacrale (c'è una violazione della funzione motoria degli arti inferiori).
- Forma bulbare (sono colpiti alcuni nuclei del cervello, il che comporta il carattere).
- Forma cervico-toracica (i sintomi primari compaiono con un cambiamento nella funzione motoria abituale arti superiori).
D'altra parte, gli esperti distinguono altri tre tipi di malattia della SLA:

Perché si verifica la SLA?
Le ragioni questa malattia purtroppo rimangono ancora poco conosciuti. Gli scienziati attualmente identificano una serie di fattori, in presenza dei quali la probabilità di ammalarsi aumenta più volte:

Malattia della SLA. Sintomi
Le foto dei pazienti con questa malattia possono essere visualizzate in libri di consultazione specializzati. Tutti loro hanno solo una cosa in comune - sintomi esterni malattia nelle fasi successive.
Per quanto riguarda i segni clinici primari di patologia, molto raramente provocano vigilanza da parte dei pazienti. Inoltre, i potenziali pazienti spesso li spiegano con lo stress costante o la mancanza di riposo dalla routine lavorativa. I seguenti sono i sintomi della malattia che compaiono nelle prime fasi:
- debolezza muscolare;
- disartria (difficoltà a parlare);
- frequenti spasmi muscolari;
- intorpidimento e debolezza degli arti;
- lievi contrazioni muscolari.
Tutti questi segni dovrebbero allertare tutti e diventare un motivo per contattare uno specialista. Altrimenti, la malattia progredirà, il che aumenta più volte la probabilità di complicanze.
Il decorso della malattia
Come si sviluppa la SLA? La malattia, i cui sintomi sono stati elencati sopra, inizia inizialmente con debolezza muscolare e intorpidimento degli arti. Se la patologia si sviluppa dalle gambe, i pazienti possono avere difficoltà a camminare, inciampare costantemente.
Se la malattia si manifesta con una violazione degli arti superiori, ci sono problemi con l'esecuzione dei compiti più elementari (abbottonarsi una camicia, girare la chiave nella serratura).
In quale altro modo si può riconoscere la SLA? Le cause della malattia nel 25% dei casi risiedono nella sconfitta del midollo allungato. Inizialmente, ci sono difficoltà con la parola e poi con la deglutizione. Tutto ciò comporta problemi con la masticazione del cibo. Di conseguenza, una persona smette di mangiare normalmente e perde peso. A questo proposito, molti pazienti cadono in uno stato depressivo, poiché la malattia di solito non colpisce
Alcuni pazienti hanno difficoltà con la formazione delle parole e persino con la normale concentrazione. Piccole violazioni di questo tipo sono spesso spiegate da una scarsa respirazione notturna. Operatori sanitari già ora dovrebbero raccontare al paziente le caratteristiche del decorso della malattia, le opzioni di trattamento, in modo che possa prendere una decisione informata sul suo futuro in anticipo.
La maggior parte dei pazienti muore per insufficienza respiratoria o polmonite. Di norma, la morte avviene entro cinque anni dalla data di conferma della malattia.
Diagnostica
Solo uno specialista può confermare la presenza di questa malattia. In questa materia, il ruolo principale è dato all'interpretazione competente del quadro clinico esistente in un particolare paziente. La diagnosi differenziale della malattia di SLA è estremamente importante.
Quale dovrebbe essere il trattamento?
Sfortunatamente, la medicina oggi non può offrire terapia efficace contro questa malattia. Come si può curare la SLA? Il trattamento dovrebbe mirare principalmente a rallentare il decorso della patologia. A tal fine vengono utilizzate le seguenti misure:

Trattamento con cellule staminali
In molti paesi europei, i pazienti con SLA vengono ora trattati con le proprie cellule staminali, il che rallenta anche la progressione della malattia. Questo tipo di terapia ha lo scopo di migliorare le funzioni primarie del cervello. Le cellule staminali trapiantate nell'area danneggiata ripristinano i neuroni, migliorano l'apporto di ossigeno al cervello e promuovono la formazione di nuovi vasi sanguigni.
L'isolamento delle stesse cellule staminali e il loro trapianto diretto, di norma, vengono effettuati in regime ambulatoriale. Dopo la terapia, il paziente rimane in ospedale per altri 2 giorni, dove gli specialisti monitorano le sue condizioni.

In questo articolo abbiamo parlato di ciò che costituisce una patologia come la SLA. Una malattia i cui sintomi potrebbero non manifestarsi per molto tempo è attualmente impossibile da curare completamente. Gli scienziati di tutto il mondo continuano a studiare attivamente questa malattia, le sue cause e la velocità di sviluppo, cercando di trovare un farmaco efficace.
Una delle malattie rare ed estremamente pericolose per la salute e la vita umana è la sclerosi laterale amiotrofica. Questa è una patologia del sistema nervoso, in cui i motoneuroni del midollo spinale, così come la corteccia e il tronco cerebrale, subiscono cambiamenti irreversibili. La malattia è cronica ed è caratterizzata da una progressione costante. Il processo patologico non è molto comune: si registrano circa 4-6 casi di malattia ogni 100.000 persone.
Dati di deviazione generali
La sclerosi laterale amiotrofica (SLA) è un disturbo del funzionamento del sistema nervoso che provoca debolezza muscolare costantemente progressiva che si manifesta in condizioni di danno selettivo dei motoneuroni. La patologia ha molti altri nomi, ad esempio la malattia di Lou Gehrig, dal nome del campione del mondo di baseball che soffriva di questa deviazione. Un altro nome per lato sclerosi amiotrofica La malattia di Charcot. È associato al nome dello psichiatra francese Jean-Martin Charcot, che descrisse la debolezza muscolare patologica nella seconda metà del XIX secolo.
Infine, la SLA, secondo l'ICD-10, è nota come malattia dei motoneuroni.

A volte, descrivendo questa condizione patologica, la parola "laterale" viene sostituita dall'epiteto "laterale". L'inclusione di questo aggettivo nel nome della malattia è dovuta al fatto che i neuroni situati nelle proiezioni laterali del midollo spinale sono più suscettibili ai cambiamenti.
I cambiamenti patologici associati alla debolezza muscolare e all'atrofia sono causati dalla distruzione dei neuroni responsabili della trasmissione del segnale dal cervello ai muscoli. Con questa malattia possono essere colpiti sia i motoneuroni centrali che quelli periferici. Il primo si trova nella corteccia cerebrale. Quando è danneggiato, si sviluppa debolezza muscolare, mentre il tono aumenta e i riflessi aumentano. Il motoneurone periferico si trova nel tronco cerebrale e oltre vari livelli midollo spinale. Se subisce cambiamenti patologici, si osserva lo sviluppo della debolezza muscolare, ma allo stesso tempo si riducono i riflessi, il tono muscolare e lo sviluppo dell'atrofia muscolare.
In condizioni di danneggiamento di uno dei motoneuroni (o di entrambi contemporaneamente), la trasmissione di un impulso da esso al muscolo è bloccata.
La sclerosi laterale amiotrofica si manifesta con un aumento della debolezza muscolare e dell'atrofia muscolare, che, di conseguenza decorso cronico, porta alla completa immobilità del paziente e alla funzionalità respiratoria compromessa.

Attualmente ci sono circa 70.000 persone con diagnosi di SLA nel mondo. Di solito questa patologia si manifesta dopo l'età di 40-50 anni. Secondo i dati clinici, di solito porta a esito letale entro 5 anni dalla diagnosi appropriata. Tuttavia, il famoso fisico Stephen Hawking convive con la malattia da oltre 50 anni.
Più di 60 mesi vive solo il 7% di tutti i pazienti.
È stato accertato che gli uomini soffrono della malattia di Charcot più spesso delle donne. Recentemente, gli scienziati lo hanno ipotizzato il numero più grande casi di patologia si registrano in persone con elevata intelligenza e atleti che non hanno avuto problemi seri con la salute.
Ragioni per lo sviluppo del processo patologico
La sclerosi laterale amiotrofica è diventata nota alla medicina non molto tempo fa e la causa principale di questa malattia non è stata ancora chiarita. Tuttavia, i ricercatori concordano sul fatto che si basa sull'accumulo di proteine patologiche insolubili nelle cellule motorie del sistema nervoso. Questo è ciò che porta alla loro morte.
Esiste una teoria secondo la quale il ruolo principale nello sviluppo di cambiamenti irreversibili appartiene a un cambiamento nelle proprietà di un enzima speciale, la cui funzione è quella di proteggere le cellule del corpo dalla distruzione dei radicali dell'ossigeno. Questo enzima è chiamato superossido dismutasi-1. Tali cambiamenti sono associati a mutazioni genetiche, che nel 25% dei casi sono ereditari. È questo fatto che determina l'ipotesi della natura genetica della SLA.
È possibile che la distruzione dei motoneuroni sia associata a mutazioni in altre strutture, ad esempio formazioni che danno alla cellula nervosa la sua forma e ne forniscono la struttura.
Quando si descrive la natura della sclerosi laterale amiotrofica, si fa menzione i seguenti motivi il suo sviluppo:
- Mutazioni geniche ereditate;
- Reazioni autoimmuni, in cui le cellule nervose del corpo vengono attaccate dal sistema immunitario;
- Il corpo produce proteine anormali che si accumulano e provocano la morte dei motoneuroni;
Presentatori del programma “Vivi sano!” parlare di malattia incurabile particolari:
- L'accumulo di acido glutammico nel corpo, che si verifica quando i processi biochimici nel corpo sono disturbati e favorisce la penetrazione del calcio nei neuroni. Questo processo porta alla formazione un largo numero radicali liberi che distruggono i motoneuroni;
- Danno virale alle cellule nervose;
- Effetto negativo sul corpo del piombo, pesticidi agricoli.
I fattori di rischio medico includono:
- Attività professionale di una persona, che comporta il contatto con piombo o altre sostanze che causano intossicazione;
- predisposizione ereditaria;
- Cambiamenti legati all'età che si verificano nel corpo;
- Fumo (nella zona di rischio speciale - fumatori con una lunga storia);
- Genere maschile.
La sclerosi laterale amiotrofica non è una malattia infettiva, non può essere contratta da un'altra persona.
Le principali forme della malattia
La malattia del motoneurone è classificata per tipo, in base alla gravità del danno alle cellule nervose. Le manifestazioni di ciascuno di essi sono sostanzialmente simili, ma, man mano che il processo patologico si sviluppa, la differenza diventa più pronunciata.
In neurologia si distinguono le seguenti forme della malattia:
- In realtà la sclerosi laterale amiotrofica. Questo nome significa la sconfitta simultanea dei neuroni del cervello e del midollo spinale. La malattia è caratterizzata debolezza generale, una sensazione di maggiore affaticamento degli arti, che si intensifica ancora di più quando si cammina o si cerca di tenere un oggetto in mano. L'aspettativa di vita media dei pazienti con una diagnosi simile va da 2 a 5 anni;
- Paralisi bulbare progressiva. Una caratteristica distintiva di questa forma è il coinvolgimento in processo patologico nervo ipoglosso e glossofaringeo, che interrompe i processi di deglutizione e il linguaggio. Rispondendo alla domanda su quanto tempo vivono i pazienti con paralisi bulbare progressiva, gli esperti notano che questo periodo è breve: da sei mesi a 3 anni;

Nella foto è la paralisi bulbare progressiva
- Sclerosi laterale primaria. Questa è una forma molto rara di patologia in cui sono interessati solo i motoneuroni del cervello. Si esprime in una sensazione di debolezza agli arti inferiori, a volte in movimenti goffi delle mani o problemi con la parola. Tale condizione non riduce l'aspettativa di vita, tuttavia c'è sempre il rischio del passaggio della malattia dalla forma della sclerosi laterale primaria alla SLA;
- Atrofia muscolare progressiva. In questo caso, c'è un danno ai motoneuroni del midollo spinale e la loro graduale morte. I muscoli diventano più deboli e di volume più piccolo, il che influisce sulla capacità motoria. L'aspettativa di vita media è di 5-10 anni.
La progressione della sclerosi laterale amiotrofica provoca complicazioni che portano alla morte del paziente. Questi dovrebbero includere:
- Disturbi respiratori. La progressiva debolezza muscolare porta alla fine alla paralisi dei muscoli coinvolti nel processo respiratorio. È questa complicazione che nella maggior parte dei casi porta alla morte;
- Difficoltà nel mangiare. Se la SLA si manifesta con il coinvolgimento dei muscoli coinvolti nel processo di deglutizione, c'è il rischio di aspirazione, che può portare a polmonite, oltre a malnutrizione e disidratazione;
- Disabilità intellettive (problemi di memoria, capacità di prendere decisioni). Molto raramente si sviluppa la demenza.
La malattia inizia con il fatto che c'è una lesione degli arti, che poi si diffonde al resto del corpo.
Quadro clinico
I primi sintomi della sclerosi laterale amiotrofica sono:
- Crampi muscolari;
- Contrazione degli arti superiori, della lingua, delle spalle;
- Sensazione di debolezza agli arti inferiori, al piede o alla caviglia;
Cari lettori, per le principali cause e sintomi della malattia, guardate il video qui sotto:
- Difficoltà a sollevare l'avampiede o le dita dei piedi dell'arto inferiore;
- Disturbi del linguaggio;
- Difficoltà a deglutire.
Con il progredire della sclerosi laterale amiotrofica, il paziente avverte rigidità muscolare, che è associata ad un aumento del tono, difficoltà nel cercare di rilassarli. Ci sono anche tali caratteristiche, come squilibrio, risate o pianti spontanei, movimenti della lingua limitati, cambio di voce.
Nelle fasi successive della malattia si verificano interruzioni della respirazione, si nota la depressione e l'incapacità di muoversi in modo indipendente.
Misure diagnostiche
La diagnosi di sclerosi laterale amiotrofica richiede un gran numero di misure specifiche. Innanzitutto, il paziente viene esaminato e intervistato da un neurologo interessato a tali informazioni:
- La gravità dei riflessi;
- Stato di coordinamento;
- Funzionalità di vista e tatto;
- Lo stato del tono muscolare e della forza muscolare.
Altri metodi che consentono di fare una diagnosi corretta includono:
- Esami del sangue per confutare gli altri probabili cause che ha provocato la comparsa di sintomi atipici;
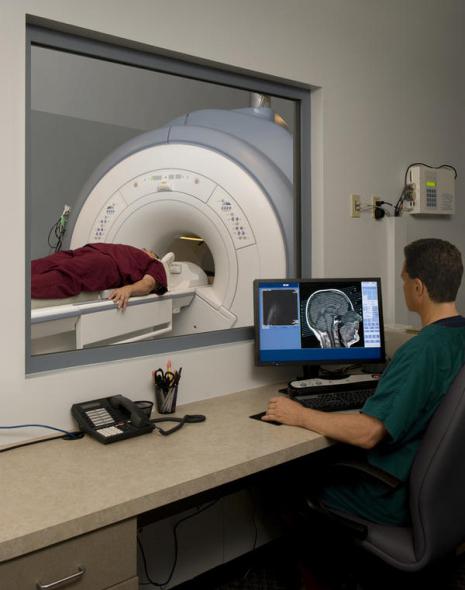
- MRI, che visualizza il tessuto nervoso e ottiene informazioni sufficienti sulla sua condizione. La tecnica conferma o esclude anche la presenza di lesioni organiche;

La procedura per eseguire una puntura lombare consiste nel perforare la membrana aracnoidea del midollo spinale con un ago di birra tra 3 e 4, o 2 e 3 vertebre lombari per la raccolta di liquori
- Puntura spinale . Lo studio del liquido cerebrospinale elimina la possibilità di sclerosi multipla in un paziente;
- Elettroneuromiografia. Usando questa procedura, vengono misurati i potenziali elettrici nei muscoli e viene anche esaminata la conduzione dell'impulso lungo le fibre nervose e il muscolo;
- Biopsia muscolare. Il campionamento di un frammento di tessuto muscolare viene eseguito se si sospetta una malattia muscolare.
Oltre a questi metodi, deve essere effettuata la diagnosi differenziale. La sclerosi laterale amiotrofica deve essere distinta da patologie come il tumore del midollo spinale, la mielopatia cervicale, la sindrome da malassorbimento, l'amiotrofia diabetica.
Esiste una cura per la SLA?
Il trattamento della sclerosi laterale amiotrofica mira a rallentare la progressione del processo patologico, poiché i cambiamenti in atto sono irreversibili. La malattia non può essere curata.
Il principale e attualmente l'unico farmaco raccomandato per rallentare la progressione della malattia è il Riluzolo o Rilutek. Il suo principale sostanza attiva previene ulteriori danni ai neuroni, rallentando così la progressione della malattia.
Altri farmaci vengono utilizzati esclusivamente per alleviare i sintomi predominanti che compromettono la qualità della vita del paziente. Questi dovrebbero includere:
- Farmaci antibatterici (in caso di infezione);
- Antidepressivi e sonniferi, necessari per gravi depressioni e disturbi del sonno;
- Steroidi anabolizzanti, che aumentano la massa muscolare;

Diversi tipi di azione dei miorilassanti
- Mezzi per ridurre l'intensità della salivazione;
- Farmaci antinfiammatori non steroidei, che possono ridurre l'intensità del dolore;
- Rilassanti muscolari per alleviare lo spasmo.
Un paziente con diagnosi di SLA necessita di dispositivi che permettano di muoversi (carrozzina, deambulatore, lettino con varie funzioni, in particolare dotato di ascensore). Affinché il paziente possa respirare, è possibile eseguire un'operazione speciale: una tracheostomia. Durante la manipolazione, viene creata chirurgicamente un'apertura nella trachea.
Al paziente deve essere garantita la piena assistenza. Particolare attenzione dovrebbe essere prestata alle misure per prevenire la formazione di piaghe da decubito. Il letto deve essere sempre asciutto e pulito, come il corpo del paziente.
Un'altra componente integrante della terapia di mantenimento per la sclerosi laterale amiotrofica è il lavoro con uno psicologo. L'aiuto di uno specialista sarà richiesto non solo per il paziente, ma anche per i suoi familiari.

A causa della disabilità nella SLA, il paziente è obbligato a utilizzare una sedia a rotelle
Previsione di questo malattia specifica deludente. La malattia progredisce costantemente, peggiorando la qualità della vita del paziente e portando infine a insufficienza respiratoria acuta e morte del paziente. A seconda della forma in cui procede il processo patologico, il paziente può vivere da 2 a 12 anni. Con la forma bulbare, così come se il paziente è dentro vecchiaia, l'aspettativa di vita è ridotta a 1-3 anni.
La sclerosi laterale amiotrofica è una malattia rara che progredisce costantemente e porta inevitabilmente alla morte del paziente. Le ragioni di questo fenomeno non sono state completamente indagate, ci sono solo ipotesi sui probabili fattori di rischio. Il trattamento della SLA è ridotto per alleviare le condizioni del paziente e fornirgli le condizioni più confortevoli per la vita.
Sclerosi laterale amiotrofica: segni, forme, diagnosi, come conviverci?
Sclerosi laterale o laterale amiotrofica (SLA), indicata come malattia del motoneurone o malattia di Charcot-Kozhevnikov, malattia del motoneurone e in alcuni luoghi il globo- La malattia di Lou Gehrig, che si applica principalmente alle regioni che parlano lingua inglese. Cari pazienti, a questo proposito, non bisogna stupirsi né dubitare se nel testo del nostro articolo si incontrano vari nomi per questo pessimo processo patologico, portando prima alla completa invalidità e poi alla morte.
Brevemente su cos'è la malattia dei motoneuroni
La base di questo terribile malattia sono lesioni del tronco encefalico che non si fermano in questa sede, ma si diffondono alle corna anteriori del midollo spinale (il livello dell'ispessimento cervicale) e al tratto piramidale, portando alla degenerazione dei muscoli scheletrici. Nelle preparazioni istologiche si trovano inclusioni citoplasmatiche chiamate corpi di Bunin e, sullo sfondo di infiltrati vascolari, si osservano cellule nervose degenerativamente alterate, rugose e morte, al posto delle quali crescono gli elementi gliali. È ovvio che il processo, oltre a tutte le parti del cervello e del midollo spinale (cervelletto, tronco, corteccia, sottocorteccia, ecc.), ai nuclei dei nervi motori cranici (nervi cranici), interessa le meningi, i vasi cerebrali e i vasi vascolari spinali letto. Il patologo dell'autopsia osserva che l'ispessimento cervicale e lombare nei pazienti diminuisce notevolmente di volume e il tronco è completamente atrofizzato.

Se anche 20 anni fa i pazienti potevano vivere a malapena 4 anni, allora ai nostri giorni c'è stato un tendenza al rialzo media durata vita, che raggiunge già 5-7 anni. La forma cerebrale non differisce ancora per la longevità (3-4 anni) e la forma bulbare non dà molte possibilità (5-6 anni). È vero, alcuni vivono per 12 anni, ma, fondamentalmente, si tratta di pazienti con una forma cervicotoracica. Tuttavia, cosa significa questo periodo se la malattia di Charcot (forme sporadiche) non risparmia l'infanzia (scuola superiore) e l'adolescenza, mentre il sesso maschile ha più “possibilità” di contrarre la malattia dei motoneuroni. I casi familiari esordiscono più spesso nell'età adulta. Pericolo reale la malattia persiste nell'intervallo tra i 40 ei 60 anni, ma dopo i 55 gli uomini non tengono più il campionato e si ammalano alla pari delle donne.
I disturbi bulbari nell'attività dei centri responsabili della funzione respiratoria e del lavoro di solito portano a un esito fatale. sistemi cardiovascolari S.
In letteratura, puoi trovare una definizione come "sindrome SLA". Questa sindrome non ha nulla a che vedere con la malattia dei motoneuroni, è causata da cause completamente diverse e accompagna altre malattie (alcune proteinemie, ecc.), anche se i sintomi della sindrome SLA sono molto simili allo stadio iniziale della malattia di Lou Gehrig, quando il clinica non ha ancora ricevuto un rapido sviluppo. Per la stessa ragione stato iniziale la sclerosi laterale amiotrofica è differenziata da () o.
Video: lezione sulla SLA dal programma educativo in neuroscienze
La forma è determinata dai sintomi predominanti
 La SLA non ha confini nel corpo umano malato, va avanti e, quindi, colpisce l'intero corpo del paziente, pertanto le forme di sclerosi laterale amiotrofica sono distinte in modo piuttosto condizionale, in base all'inizio del processo patologico e altro segni luminosi sconfitta. Esattamente prevalente i sintomi durante la sclerosi laterale amiotrofica e le aree colpite non isolate consentono di determinarne le forme, che possono essere rappresentate come segue:
La SLA non ha confini nel corpo umano malato, va avanti e, quindi, colpisce l'intero corpo del paziente, pertanto le forme di sclerosi laterale amiotrofica sono distinte in modo piuttosto condizionale, in base all'inizio del processo patologico e altro segni luminosi sconfitta. Esattamente prevalente i sintomi durante la sclerosi laterale amiotrofica e le aree colpite non isolate consentono di determinarne le forme, che possono essere rappresentate come segue:
- cervicotoracico, che prima di tutto iniziano a sentire le mani, la zona delle scapole, l'intero cingolo scapolare. È molto difficile per una persona controllare i movimenti, sui quali, prima della malattia, non era necessario concentrare nemmeno l'attenzione. I riflessi fisiologici aumentano, i riflessi patologici sorgono in parallelo. Poco dopo che le mani cessano di obbedire, si verifica l'atrofia muscolare della mano ("zampa di scimmia") e il paziente in quest'area viene immobilizzato. Divisioni inferiori inoltre non si fanno da parte e sono coinvolti nel processo patologico;
- lombosacrale. Come le mani, gli arti inferiori iniziano a soffrire, appare la debolezza dei muscoli degli arti inferiori, accompagnata da contrazioni, spesso convulsioni, quindi si verifica l'atrofia muscolare. I riflessi patologici (segno positivo di Babinsky, ecc.) Sono criteri diagnostici, poiché si osservano già all'inizio della malattia;
- forma bulbare- uno dei più gravi, che solo in rari casi consente al paziente di allungare l'aspettativa di vita di oltre 4 anni. Oltre ai problemi con la parola ("nasale") e alle espressioni facciali incontrollabili, ci sono segni di difficoltà a deglutire, che si trasformano in una completa incapacità di mangiare da soli. Il processo patologico, che copre l'intero corpo del paziente, ha un effetto molto negativo sulle capacità funzionali dei sistemi respiratorio e cardiovascolare, quindi le persone con questa forma muoiono prima che si sviluppino paresi e paralisi. Non ha senso tenere un paziente del genere su un ventilatore per molto tempo ( ventilazione artificiale polmoni) e alimentarsi con contagocce e con l'ausilio di una gastrostomia, poiché la percentuale di speranza di guarigione in questa forma è praticamente ridotta a zero;
- cerebrale che si chiama alto. È noto che tutto viene dalla testa, quindi non sorprende che nella forma cerebrale sia le braccia che le gambe siano colpite e atrofizzate. Inoltre, è molto comune che un paziente con questa variante pianga o rida senza motivo. Queste azioni, di regola, non sono collegate alle sue esperienze ed emozioni. Dopotutto, se un paziente piange nelle sue condizioni, questo può essere compreso, ma è improbabile che diventi divertente dalla sua malattia, quindi possiamo dire che tutto accade spontaneamente, indipendentemente dal desiderio della persona. Secondo la gravità del decorso, la forma cerebrale non è praticamente inferiore a quella bulbare, che porta anche rapidamente alla morte del paziente;
- polinevrotico(poli significa molto). La forma si manifesta con lesioni multiple dei nervi e atrofia muscolare, paresi e paralisi degli arti. Molti autori non lo distinguono in una forma separata e, in generale, la classificazione di diversi paesi o autori diversi può differire, in cui non c'è nulla di vergognoso, quindi non dovresti concentrarti su questo, inoltre, cerebrale e forma bulbare non bypassa nessuna fonte.
Cause di malattia...
I fattori che possono innescare questo grave processo patologico non sono così numerosi, ma una persona può incontrarne uno ogni giorno, indipendentemente dall'età, dal sesso e posizione geografica, salvo, ovviamente, la predisposizione ereditaria, che è caratteristica solo per una certa parte della popolazione (5-10%).
Quindi, le cause della malattia del motoneurone:
- Intossicazione (qualsiasi, ma soprattutto - sostanze industria chimica, dove il ruolo principale è assegnato all'influenza dei metalli: alluminio, piombo, mercurio e manganese);
- Malattie infettive causate dall'attività vitale di vari virus nel corpo umano. Qui, un posto speciale spetta a una lenta infezione causata da un virus neurotropico fino ad allora sconosciuto;
- lesioni elettriche;
- Mancanza di vitamine (ipovitaminosi);
- La gravidanza può provocare una malattia dei motoneuroni;
- Neoplasie maligne (soprattutto cancro ai polmoni);
- Operazioni (rimozione di parte dello stomaco);
- Predisposizione genetica programmata (casi familiari di malattia del motoneurone). Il colpevole della sclerosi laterale amiotrofica è un gene mutato situato sul cromosoma 21, che è trasmesso prevalentemente per via ereditaria autosomica dominante, sebbene in alcuni casi si verifichi anche una variante autosomica recessiva, sebbene in misura minore;
- Motivo inspiegabile.
…e le sue manifestazioni cliniche
I sintomi della sclerosi laterale amiotrofica sono caratterizzati, innanzitutto, dalla comparsa di paresi periferica e centrale delle mani, come indicato dai seguenti segni:
- I riflessi periostale e tendineo iniziano a mostrare una tendenza ad aumentare;
- Atrofia dei muscoli delle mani e della zona scapolare;
- Il verificarsi di riflessi patologici (sintomi superiori di Rossolimo, che si riferisce ai riflessi patologici della mano, un sintomo positivo di Babinsky, ecc.);
- Stop clono, aumento dei riflessi di Achille e del ginocchio;
- La comparsa di contrazioni fibrillari dei muscoli del cingolo scapolare e, inoltre, dei muscoli delle labbra e della lingua, che possono essere facilmente visti se si colpisce il muscolo nell'area interessata con il martello di un neurologo;
- La formazione di paralisi bulbare, che si manifesta con soffocamento, disartria, raucedine, abbassamento della mascella (inferiore, ovviamente), eccessiva salivazione;
- Con la malattia dei motoneuroni, la psiche umana praticamente non soffre, tuttavia, è improbabile che una patologia così grave non influisca in alcun modo sull'umore e non influisca sul background emotivo. Di norma, i pazienti in tali situazioni cadono profonda depressione, perché sanno già qualcosa della loro malattia e la condizione dice molto;

Ovviamente, coinvolgendo l'intero organismo nel processo, la malattia di Charcot dà una sintomatologia ricca e diversificata, che, però, può essere sinteticamente rappresentata da sindromi:
- Lento e paralisi spastica mani e piedi;
- Atrofia muscolare con:
a) contrazioni fibrillari causate dall'irritazione delle corna anteriori del midollo spinale, che portano all'eccitazione di alcune fibre muscolari (individuali);
b) contrazioni fasciali causate dal movimento di un intero fascio di muscoli e derivanti dall'irritazione delle radici; - Sindrome dei disturbi bulbari.
I principali criteri diagnostici sono i riflessi e l'ENMG
Per quanto riguarda la diagnosi, dipende principalmente dallo stato neurologico, ma il principale metodo strumentale per la ricerca della SLA si riconosce l'ENMG (elettroneuromiografia), il resto dei test viene effettuato per escludere patologie simili nei sintomi o per studiare il corpo del paziente, in particolare lo stato dell'apparato respiratorio e dell'apparato muscolo-scheletrico. Pertanto, l'elenco degli studi necessari include:
- Analisi cliniche generali (tradizionali) ( analisi generali sangue e urina);
- LHC (biochimica);
- Puntura spinale (piuttosto da escludere sclerosi multipla, poiché non ci sono cambiamenti nel liquido cerebrospinale nella malattia di Charcot);
- biopsia muscolare;
- esame R-grafico;
- MRI per rilevare o escludere lesioni organiche;
- Spirogramma (studio della funzione respirazione esterna), che è molto importante per tali pazienti, dato che la funzione respiratoria spesso soffre nella sclerosi laterale amiotrofica.
Per mantenere e prolungare la vita
La terapia per la malattia del motoneurone è principalmente Ha lo scopo di rafforzare in generale, mantenere il corpo e alleviare i sintomi. Man mano che il processo patologico si sviluppa, l'insufficienza respiratoria aumenta, quindi il paziente, al fine di migliorare l'attività respiratoria, prima (mentre è ancora su una sedia a rotelle) passa al dispositivo NIVL (per la ventilazione polmonare non invasiva), quindi, quando non può far fronte più lungo, ad apparecchiature di ventilazione fisse.

Davvero un trattamento efficace per la sclerosi laterale amiotrofica non è stato ancora inventato, tuttavia, è ancora necessario essere curato e al paziente viene prescritta una terapia farmacologica:
- Rilutek (Riluzolo) è l'unico farmaco mirato disponibile. Appena un po' (circa un mese) allunga la vita e permette di allungare i tempi prima di trasferire il paziente in ventilazione meccanica;
- Usato per migliorare la parola e la deglutizione farmaci anticolinesterasici(galantamina, prozerina);
- Elenium, sibazon (diazepam), miorilassanti aiutano ad alleviare gli spasmi;
- Con depressione e disturbi del sonno - tranquillanti, antidepressivi e sonniferi;
- quando complicazioni infettive tenuto terapia antibiotica(antibiotici);
- Per il dolore vengono utilizzati FANS (antinfiammatori non steroidei) e analgesici e successivamente il paziente viene trasferito ad antidolorifici narcotici;
- L'amitriptilina è prescritta per ridurre la salivazione;
- La composizione del trattamento, di regola, comprende anche le vitamine del gruppo B, steroide anabolizzante, contribuendo all'aumento massa muscolare(riabolire), farmaci nootropi(piracetam, cerebrolysin, nootropil).
Una buona cura migliora la qualità della vita
Difficilmente si può discutere con l'affermazione che un paziente con la malattia di Charcot ha bisogno cura speciale. È in un modo speciale, perché una poppata vale qualcosa. E la lotta alle piaghe da decubito? E la depressione? Il paziente è critico nei confronti delle sue condizioni, è molto preoccupato che ogni giorno le sue condizioni peggiorino e, alla fine, smette (contro la sua volontà) di servirsi, non può comunicare con gli altri e godersi una cena deliziosa.
Un tale paziente ha bisogno di:
- In un letto funzionale dotato di ascensore,
- In una poltrona con attrezzatura che sostituisce un wc;
- In carrozzina comandata da pulsanti che il paziente può ancora manovrare;
- Nei mezzi di comunicazione, per i quali il laptop è più adatto.
La prevenzione delle piaghe da decubito è molto importante. In questi casi, non si fanno aspettare a lungo, quindi il letto dovrebbe essere pulito e asciutto, così come il corpo del paziente.
Il paziente mangia prevalentemente cibi liquidi, ben ingeriti, ricchi di proteine e vitamine (purché sia preservata la funzione di deglutizione). Successivamente, il paziente viene alimentato attraverso una sonda e quindi ricorre a una misura forzata, ma ultima: l'imposizione gastrostomia.

Ovviamente, un paziente con sclerosi laterale amiotrofica soffre molto: sia moralmente che fisicamente. Allo stesso tempo, soffrono anche le persone che si prendono cura di lui, per le quali è una persona vicina. D'accordo, è molto difficile guardare negli occhi sbiaditi, vedere dolore e disperazione e non essere in grado di aiutare a sconfiggere la malattia, curare, riportare in vita. cara persona. I parenti che si prendono cura di una persona così malata perdono forza e spesso si scoraggiano e si deprimono, e quindi hanno anche bisogno dell'aiuto di uno psicoterapeuta con la nomina di sedativi e antidepressivi.
Di solito, nella descrizione del trattamento di qualsiasi malattia, i lettori cercano misure preventive e modi per sbarazzarsi di un particolare disturbo. rimedi popolari. Veramente consigliato medicina alternativa contenente grandi quantità di vitamine del gruppo B, grano germogliato e chicchi di avena, Noci e la propoli, forse, non interferirà con il paziente, ma non lo cureranno nemmeno. Inoltre, non dobbiamo dimenticare che queste persone hanno spesso problemi con la deglutizione, quindi nel caso della malattia di Charcot, non dovresti fare affidamento sulla medicina tradizionale.
Ecco cos'è: la sclerosi laterale amiotrofica (e molti altri suoi nomi). La malattia è terribilmente insidiosa, incomprensibile e incurabile. Forse un giorno una persona sarà in grado di domare questa malattia, secondo almeno speriamo per il meglio, perché gli scienziati di tutto il mondo stanno lavorando su questo problema.
Video: ALS nel programma "Live Healthy!"
La sindrome SLA (sclerosi laterale amiotrofica) è una delle rare malattie neurologiche. Secondo le statistiche, la frequenza della patologia è di 3 persone su 100 mila La formazione di anomalie degenerative-distrofiche è dovuta alla morte degli assoni nervosi, lungo i quali gli impulsi vengono trasmessi a cellule muscolari. Un processo anormale di distruzione dei neuroni si verifica nella corteccia cerebrale, le corna (anteriore) del midollo spinale. A causa della mancanza di innervazione, la contrazione muscolare si interrompe, si sviluppa atrofia, paresi.
Jean-Martin Charcot è stato il primo a descrivere la malattia, dando il nome di "sclerosi amiotrofica laterale (laterale) (SLA)." Sulla base dei risultati dello studio, ha concluso che nella maggior parte dei casi l'eziologia è sporadica. Nel 10% dei pazienti, la causa era una predisposizione ereditaria. Si sviluppa principalmente dopo i 45 anni, nelle donne è meno comune che negli uomini. Il secondo nome - sindrome di Lou Gehrig - è comune nei paesi di lingua inglese, è stata assegnata un'anomalia in onore del famoso giocatore di baseball, che ha concluso la sua carriera su una sedia a rotelle a causa di una malattia all'età di 35 anni.
Classificazione e caratteristiche
La classificazione della patologia dipende dalla posizione della lesione. Due tipi di neuroni sono coinvolti nell'attività motoria: quello principale, situato negli emisferi cerebrali, e quello periferico, situato a livelli diversi. colonna vertebrale. Quello centrale invia un impulso al secondario e, a sua volta, alle cellule dei muscoli scheletrici. L'aspetto della SLA sarà diverso a seconda del centro in cui è bloccata la trasmissione dai motoneuroni.
Sul fase iniziale Il decorso clinico dei sintomi è lo stesso indipendentemente dal tipo: spasmi, intorpidimento, ipotensione muscolare, debolezza delle braccia e delle gambe. Per sintomi generali si applica a:
- Comparsa episodica di crampi (contrazioni dolorose) sulla zona interessata.
- La graduale diffusione dell'atrofia in tutte le parti del corpo.
- Disturbo della funzione motoria.
I tipi della malattia procedono senza perdita di riflessi sensibili.
forma lombosacrale
È una manifestazione di mielopatia (distruzione del midollo spinale), causata dalla morte dei neuroni periferici situati nella colonna sacrale (corna anteriori). La sindrome SLA è accompagnata da sintomi:
- Debolezza di uno, poi di entrambi gli arti inferiori.
- Mancanza di riflessi tendinei.
- La formazione dell'atrofia muscolare iniziale, determinata visivamente da una diminuzione della massa ("restringimento").
- Fascicolazioni ondulate.
Il processo coinvolge gli arti superiori con le stesse manifestazioni.
forma cervicotoracica
La sindrome è caratterizzata dalla morte degli assoni dei neuroni secondari situati in sezione superiore colonna vertebrale, che porta alla manifestazione dei sintomi:
- diminuzione del tono in una mano, dopo un periodo di tempo il processo patologico si diffonde nella seconda;
- si nota atrofia muscolare, accompagnata da paresi, fascicolazione;
- le falangi si deformano, assumendo l'aspetto di una "spazzola di scimmia";
- si manifestano i segni del piede, caratterizzati da un cambiamento nella funzione motoria, l'atrofia muscolare è assente.
Sintomo di sconfitta cervicaleè una testa costantemente inclinata in avanti.

forma bulbare
Questo tipo di sindrome è caratterizzato da un decorso clinico grave, i motoneuroni muoiono nella corteccia cerebrale. L'aspettativa di vita dei pazienti con questo modulo non supera i cinque anni. Il debutto è accompagnato da:
- violazione della funzione articolatoria, apparato vocale;
- fissazione della lingua in una certa posizione, è difficile per loro muoversi, si notano contrazioni ritmiche;
- spasmi involontari dei muscoli mimici;
- disfunzione della deglutizione dovuta a spasmi nell'esofago.
La progressione della sclerosi laterale amiotrofica di tipo bulbare forma una completa atrofia dei muscoli facciali e del collo. Il paziente non può aprire autonomamente la bocca per mangiare, le capacità comunicative sono perse, la capacità di pronunciare chiaramente le parole. Aumento del riflesso del vomito e della mascella. Spesso la malattia si verifica sullo sfondo di risate involontarie o lacrimazione.
forma alta
Questo tipo di SLA inizia con un danno ai neuroni centrali, nel processo di sviluppo copre quelli periferici. I pazienti con una forma elevata della sindrome non vivono fino allo stadio della paralisi, perché i muscoli del cuore e degli organi respiratori muoiono rapidamente e si formano ascessi nelle aree interessate. Una persona non può muoversi in modo indipendente, l'atrofia copre l'intero muscolo scheletrico. La paresi porta a defecazione e minzione incontrollate.
La condizione è aggravata dalla costante progressione della sindrome, nella fase terminale l'atto respiratorio è impossibile, è necessaria la ventilazione dei polmoni mediante un apposito apparato.
Cause
La sindrome della sclerosi laterale amiotrofica nella maggior parte dei casi procede con una genesi incerta. Nel 10% dei pazienti con questa diagnosi, la causa dello sviluppo è stata la trasmissione di un gene mutato della generazione precedente in modo autosomico dominante. L'eziologia della formazione della malattia può essere una serie di fattori:
- Lesione infettiva del cervello, midollo spinale con un virus neurotropico stabile poco studiato.
- Assunzione insufficiente di vitamine (ipovitaminosi).
- La gravidanza può provocare la sindrome della SLA nelle donne.
- Crescita delle cellule tumorali nei polmoni.
- Bypass gastrico.
- Forma cronica di osteocondrosi della regione cervicale.
Il gruppo di rischio comprende le persone che sono costantemente in contatto con i concentrati sostanze chimiche, metalli pesanti (piombo, mercurio).
Studi diagnostici
L'esame prevede la differenziazione della sindrome SLA dalla malattia SLA. La patologia indipendente procede senza violazione degli organi interni, capacità mentale, riflessi sensibili. Per un trattamento adeguato, è necessario escludere malattie con sintomi simili mediante la diagnostica:
- amiotrofie craniovertebrali spinali;
- effetti residui della poliomielite;
- linfoma maligno;
- paraproteinemia;
- endocrinopatia;
- mielopatia cervicale con sindrome SLA.

Le misure diagnostiche per determinare la malattia includono:
- radiografia del torace;
- risonanza magnetica del midollo spinale, cervello;
- elettrocardiogrammi;
- elettroneurografia;
- studi sul livello di funzionamento della tiroide;
- puntura cerebrospinale, lombare;
- analisi genetiche per rilevare le mutazioni;
- spirogrammi;
- studio di laboratorio nel sangue di proteine, VES, creatinfosfochinasi, urea.
Trattamenti efficaci
È impossibile eliminare completamente la malattia, in Russia non esiste un farmaco brevettato in grado di fermare lo sviluppo clinico. Nei paesi europei, il riluzolo viene utilizzato per rallentare la diffusione dell'atrofia muscolare. Il compito del farmaco è di inibire la produzione di glutammato, un'alta concentrazione del quale danneggia i neuroni cerebrali. I test hanno dimostrato che i pazienti che assumono il farmaco vivono un po' più a lungo, ma muoiono comunque per insufficienza respiratoria.
Il trattamento è sintomatico, il compito principale della terapia è mantenere la qualità della vita, prolungare la capacità di cura di sé. Nel processo di sviluppo della sindrome, i muscoli degli organi responsabili dell'atto respiratorio vengono gradualmente colpiti. La carenza di ossigeno è compensata dal BIPAP, apparato IPPV utilizzato di notte. L'attrezzatura facilita le condizioni del paziente, è facile da usare e utilizzata a casa. Dopo la completa atrofia dell'apparato respiratorio, il paziente viene trasferito in un apparato di ventilazione polmonare stazionario (NIVL).
Il trattamento conservativo dei sintomi contribuisce a:
- Sollievo dalle convulsioni "Carbamazepine", "Tizanil", "Phenytoin", "Isoptin", "Baclofen", iniezione di chinino solfato.
- Normalizzazione processi metabolici nella massa muscolare con agenti anticolinesterasici ("Berlition", "Espa-Lipon", "Glutoxim", Acido lipoico, "Cortexin", "Elkar", "Levokarnitin", "Prozerin", "Kalimin", "Pyridostigmin", " Milgamma", "Thiogamma", Vitamine del gruppo B A, E, C).
- Rimozione di fascicolazioni (Elenium, Sirdalud, Sibazon, Diazepam, Mydocalm, Baklosan).
- Miglioramento della funzione di deglutizione ("Prozerin", "Galantamina").
- eliminazione sindrome del dolore analgesico "Fluoxetina" con successivo trasferimento del paziente alla morfina.
- Normalizzazione della quantità di saliva secreta da Buscopan.
- Aumenta la massa muscolare "Retabolil".
- Rimozione dei disturbi mentali con antidepressivi (Paxil, Sertralina, Amitriptilina, Fluoxetina).
Se necessario, viene prescritta una terapia antibiotica con gli antibiotici Fluorochinolo, Cefalosporina, Carbapenem. Inoltre, il corso del trattamento include farmaci nootropici: Nootropil, Piracetam, Cerebrolysin.
I pazienti con sindrome SLA necessitano di dispositivi speciali per semplificare la vita, tra cui:
Discutiamo in dettaglio di una malattia come la sclerosi laterale amiotrofica. Scopri di cosa si tratta, quali sono i sintomi e le cause. Tocchiamo la diagnosi e il trattamento della malattia di SLA. E ce ne saranno molti altri consiglio utile su questo argomento.
è una malattia neurodegenerativa accompagnata dalla morte dei motoneuroni centrali e periferici. Questo porta a una graduale atrofia dei muscoli scheletrici, disfagia, disartria, insufficienza alimentare e respiratoria. La malattia progredisce costantemente ed è accompagnata dalla morte.
Questo determina la rilevanza dello studio del problema. Fu descritto per la prima volta dallo psichiatra francese Jean-Martin Charcot nel 1869. Per il quale ha ricevuto un tale secondo nome come La malattia di Charcot.
 Jean Martin Charcot
Jean Martin Charcot Negli Stati Uniti e in Canada, è anche conosciuto come La malattia di Lou Gehrig. Per 17 anni è stato un giocatore di baseball americano di prim'ordine. Ma sfortunatamente, all'età di 36 anni, si ammalò di sclerosi laterale amiotrofica. E l'anno successivo morì.

È noto che la maggior parte dei pazienti con SLA sono persone con un elevato potenziale intellettuale e professionale. Diventano rapidamente gravemente disabili e muoiono.
L'analizzatore motore è interessato. Questo fa parte del sistema nervoso. Trasmette, raccoglie ed elabora le informazioni dai recettori del sistema muscolo-scheletrico. Organizza anche movimenti umani coordinati.
Se osservi la figura sottostante, vedrai che il sistema di propulsione è organizzato in modo molto complesso.
 La struttura dell'analizzatore motorio
La struttura dell'analizzatore motorio Alla destra angolo superiore vediamo la corteccia motoria primaria, il tratto piramidale che va al midollo spinale. Sono queste strutture che sono colpite nella SLA.
Anatomia del tratto piramidale
Ecco l'anatomia del tratto piramidale. Qui vedete un'area motoria aggiuntiva, la corteccia premotoria.

Queste trasformazioni trasmettono segnali dal cervello ai motoneuroni del midollo spinale. Innervano i muscoli scheletrici e regolano i movimenti volontari.
La sclerosi laterale amiotrofica è una malattia neurodegenerativa insolita! Codice ICD 10— G12.2.
Tutto il peggio con lui succede quando una persona continua a non provare nulla. In questa fase preclinica Il 50 - 80% dei motoneuroni muore dopo che un fallimento genetico si realizza con la partecipazione di fattori ambiente esterno. Quindi, quando rimane il 20% dei motoneuroni resistenti, inizia la malattia stessa.
 Manifestazione della sclerosi laterale amiotrofica
Manifestazione della sclerosi laterale amiotrofica La patogenesi della sclerosi laterale amiotrofica
Se parliamo della patogenesi della sclerosi laterale amiotrofica, prima di tutto dobbiamo capire quanto segue. Ci sono vari fattori genetici (in gran parte sconosciuti).
Si realizzano in condizioni di vulnerabilità selettiva dei motoneuroni. Cioè, in quelle condizioni che assicurano il normale lavoro vitale e fisiologico di queste cellule.
 La patogenesi della sclerosi laterale amiotrofica
La patogenesi della sclerosi laterale amiotrofica Tuttavia, nel condizioni patologiche svolgono un ruolo nello sviluppo della degenerazione. Ciò porta successivamente ai principali meccanismi di patogenesi.
Motoneuroni- Queste sono le cellule più grandi del sistema nervoso con lunghi processi di conduzione ( fino a 1 metro). Hanno bisogno di alti costi energetici.
 Motoneuroni
Motoneuroni Ciascuno di questi motoneuroni è una centrale elettrica speciale. Lei prende il sopravvento grande quantità impulsi e poi li trasmette per attuare movimenti umani coordinati.
Queste cellule hanno bisogno di molto calcio intracellulare. È lui che assicura il lavoro di molti sistemi di motoneuroni. Pertanto, la produzione di proteine che legano il calcio è ridotta nelle cellule.
L'espressione di alcuni recettori del glutammato (ampa) e l'espressione di proteine (bcl-2) che impediscono la morte programmata di queste cellule è ridotta.
In condizioni patologiche, queste caratteristiche del motoneurone agiscono sul processo di degenerazione. Di conseguenza, c'è:
- Tossicità (eccitotossicità da glutammato) aminoacidi stimolanti
- Stress ossidativo (ossidativo).
- Interruzione del citoscheletro dei motoneuroni
- La degradazione delle proteine è disturbata dalla formazione di alcune inclusioni
- Esiste un effetto citotossico delle proteine mutanti (sod-1)
- Apoptosi o morte cellulare programmata dei motoneuroni
Tipi di malattie
SLA familiare(Fals) - si verifica quando il paziente nella storia della famiglia ha casi simili di questa malattia. Costituisce il 15%.

In altri casi, quando hanno percorsi di eredità più complessi (85%), si parla di SLA sporadica.
Epidemiologia della malattia del motoneurone
Se parliamo dell'epidemiologia della malattia dei motoneuroni, il numero di nuovi pazienti all'anno è di circa 2 casi ogni 100.000 persone. La prevalenza (il numero di pazienti con SLA contemporaneamente) varia da 1 a 7 casi ogni 100.000 persone.

Di norma, le persone tra i 20 e gli 80 anni si ammalano. Sebbene siano possibili eccezioni.
aspettativa di vita media:
- se la malattia di SLA inizia con un disturbo del linguaggio (con un esordio bulbare), di solito vivono 2,5 anni
- se inizia con qualche tipo di disturbo motorio (esordio spinale), allora sono 3,5 anni
Tuttavia, va notato che il 7% dei pazienti vive più di 5 anni.
Loci genetici della SLA familiare
Qui vediamo molti tipi di SLA familiare. Sono state scoperte più di 20 mutazioni. Alcuni di loro sono rari. Alcuni sono comuni.
Loci genetici della SLA familiare
| Tipo di | Frequenza | Gene | Clinica |
|---|---|---|---|
| FALS1 (21q21) | 15-20% FALSI | SOD-1 | Tipico |
| FALS2 (2q33) | Raro, AP | Alsin | Atipico, SE |
| FALS3 (18q21) | Una famiglia | sconosciuto | Tipico |
| FALS4 (9q34) | Molto raro | sentaxin | Atipico, SE |
| FALS5 (15q15) | Raro, AR | sconosciuto | Atipico, SE |
| FALS6 (16q12) | 3-5% FALSI | FUS | Tipico |
| FALS7 (20p13) | Una famiglia | ? | Tipico |
| FALS8 | Molto raro | VAP | Atipico, diverso. |
| FALS9 (14q11) | Raro | Angiogenina | Tipico |
| FALS10 (1p36) | 1-3% falso fino al 38% familiare e 7% sporadico |
TDP-43 | Tipico ALS, FTD, ALS-FTD |
Ci sono anche nuovi geni per la SLA. Non ne abbiamo mostrati alcuni. Ma la linea di fondo è che tutte queste mutazioni portano a un percorso finale. Allo sviluppo del danno ai motoneuroni centrali e periferici.
Classificazione delle malattie dei motoneuroni
Di seguito è riportata una classificazione delle malattie dei motoneuroni.
Classificazione Norris (1993):
- Sclerosi laterale amiotrofica - 88% dei pazienti:
- debutto bulbare della SLA - 30%
- debutto al petto - 5%
- diffuso - 5%
- cervicale - 40%
- lombare - 10%
- respiratorio - meno dell'1%
- Paralisi bulbare progressiva - 2%
- Atrofia muscolare progressiva - 8%
- Sclerosi laterale primaria - 2%
Bass Variations di Hondkarian (1978):
- Classico - 52% (quando le lesioni dei motoneuroni centrali e periferici sono rappresentate uniformemente)
- Segmentale-nucleare - 32% (piccoli segni di lesione centrale)
- Piramidale - 16% (vediamo segni di una lesione periferica non brillanti come i segni di una centrale)
Patomorfosi naturale
Se parliamo di debutti diversi, allora va notato che la sequenza di sviluppo dei sintomi è sempre certa.
 Patomorfosi nell'esordio bulbare e cervicale della SLA
Patomorfosi nell'esordio bulbare e cervicale della SLA In debutto bulbare prima ci sono i disturbi del linguaggio. Poi problemi di deglutizione. Poi c'è la paresi negli arti e disturbi respiratori.
In debutto cervicale il processo di violazione inizia con una mano e poi passa nell'altra. Successivamente, possono verificarsi disturbi bulbari e disturbi del movimento nelle gambe. Tutto inizia dal lato su cui ha sofferto la mano primaria.
Se parlare esordio toracico della SLA, quindi il primo sintomo che i pazienti di solito non notano è la debolezza dei muscoli della schiena. Lo stato è rotto. Quindi si verifica la paresi nel braccio insieme all'atrofia.
 Patomorfosi nell'esordio toracico e lombare della SLA
Patomorfosi nell'esordio toracico e lombare della SLA In debutto lombare una gamba è colpita per prima. Quindi viene catturato il secondo, dopo di che la malattia passa alle mani. Poi ci sono i disturbi respiratori e bulbari.
Manifestazioni cliniche della malattia di Charcot
Le manifestazioni cliniche della malattia di Charcot includono:
- Segni di danno ai motoneuroni periferici
- Segni di danno ai motoneuroni centrali
- Combinazione di sindromi bulbari e pseudobulbari
Complicanze fatali che portano alla morte:
- Disfagia (deglutizione ridotta) e malnutrizione alimentare (nutrizionale).
- Disturbi delle vie respiratorie della colonna vertebrale e dello stelo per atrofia dei muscoli respiratori principali e accessori
Segni di danno al motoneurone centrale
I segni di danno al motoneurone centrale includono:
- Perdita di destrezza: la malattia inizia con il fatto che una persona inizia ad avere difficoltà ad allacciare bottoni, legare i lacci delle scarpe, suonare il piano o infilare un ago
- Quindi la forza muscolare diminuisce
- Aumento del tono muscolare spastico
- Compare l'iperreflessia
- Riflessi patologici
- Sintomi pseudobulbari
Segni di danno al motoneurone periferico
Segni di danno al motoneurone periferico sono combinati con segni di danno al centro:
- Fascicolazioni (contrazioni visibili nei muscoli)
- Crampi (crampi muscolari dolorosi)
- Paresi e atrofia dei muscoli scheletrici della testa, del tronco e degli arti
- Ipotensione muscolare
- iporeflessia
Sintomi costituzionali della SLA
I sintomi costituzionali della SLA includono:
- Cachessia associata alla SLA(perdita di oltre il 20% del peso corporeo in 6 mesi) è un evento catabolico nel corpo. È associato alla morte di un gran numero di cellule del sistema nervoso. In questo caso, ai pazienti vengono prescritti ormoni anabolizzanti. Anche la cachessia può svilupparsi dalla malnutrizione.
- Fatica(ristrutturazione delle placche terminali) - in un certo numero di pazienti è possibile un decremento del 15 - 30% con EMG

Sintomi rari nella sclerosi laterale amiotrofica
Ecco a cosa si applica sintomi rari nella sclerosi laterale amiotrofica:
- Disturbi sensoriali. Tuttavia, è stato dimostrato che nel 20% dei casi i pazienti con SLA (soprattutto negli anziani) presentano disabilità sensoriali. Questa è polineuropatia. Inoltre, se la malattia inizia con le mani, il paziente le appende semplicemente. Hanno una circolazione ridotta. Anche i potenziali possono diminuire in questi nervi sensoriali.
- La violazione delle funzioni oculomotorie, della minzione e delle feci è estremamente rara. Meno dell'1%. Tuttavia, sono possibili disturbi secondari più frequenti. Questa è una debolezza dei muscoli del pavimento pelvico.
- La demenza (demenza) si verifica nel 5% dei casi.
- Compromissione cognitiva - 40%. Nel 25% dei casi sono progressivi.
- Piaghe da decubito - meno dell'1%. Di norma, si verificano con grave malnutrizione alimentare.

Vale la pena dire che in presenza di questi sintomi si può dubitare della diagnosi.
Tuttavia, va sempre ricordato che se un paziente ha un quadro clinico tipico di questa malattia e presenta questi sintomi, allora può essere fatta una diagnosi di SLA con caratteristiche.
Deterioramento cognitivo moderato e demenza
Parliamo di più del lieve deterioramento cognitivo e della demenza nella malattia di Lou Gehrig. Qui vediamo spesso mutazioni genetiche C9orf72. Porta allo sviluppo della SLA, della demenza frontotemporale e della loro combinazione.
Ci sono tre opzioni per lo sviluppo di questo disturbo:
- variante comportamentale- questo è quando la motivazione del paziente diminuisce (sindrome apato-abulica). O viceversa, c'è disinibizione. La capacità di una persona di comunicare attivamente e adeguatamente nella società è ridotta. Le critiche sono in calo. Il flusso della parola è alterato.
- esecutivo- Violazione della pianificazione dell'azione, generalizzazione, fluidità del discorso. I processi logici sono interrotti.
- Semantico (discorso)- Raramente si manifesta un discorso fluido e privo di significato. Tuttavia, si verificano spesso disnomia (dimenticano le parole), parafasie fonemiche (danni alle zone vocali frontali). Spesso commettono errori grammaticali e balbettano. C'è un paragrafo (disordine scrivere) e aprassia orale (non è possibile avvolgere le labbra attorno al tubo dello spirografo). C'è anche dislessia e disgrafia.
Criteri diagnostici per la disfunzione frontotemporale secondo D. Neary (1998)
I criteri diagnostici per la disfunzione frontotemporale secondo D. Neary includono caratteristiche obbligatorie come:
- Esordio insidioso e progressione graduale
- Perdita precoce dell'autocontrollo del comportamento
- Il rapido emergere delle difficoltà di interazione nella società
- Appiattimento emotivo nelle prime fasi
- Precoce declino delle critiche
La diagnosi non contraddice il fatto che un tale disturbo possa manifestarsi prima dei 65 anni. La diagnosi è messa in discussione quando il paziente abusa di alcol. Se tali disturbi sono divorziati in modo acuto, sono stati preceduti da un trauma cranico, la diagnosi di disfunzione frontotemporale viene rimossa.
Il paziente è mostrato di seguito. Ha un sintomo di occhi vuoti. Questo non è un sintomo specifico. Ma con la SLA, quando una persona non può parlare o muoversi, questo è qualcosa a cui prestare attenzione.
 Sintomo di "occhio vuoto" in un paziente con SLA + paragrafo FTD e "stile telegrafico" in FTD
Sintomo di "occhio vuoto" in un paziente con SLA + paragrafo FTD e "stile telegrafico" in FTD A destra c'è un esempio in cui il paziente scrive in stile telegrafico. Scrive singole parole e commette errori.
Guarda quanto è spessa la pelle nella SLA. È difficile per i pazienti perforare la pelle con un elettrodo ad ago. Inoltre, ci sono difficoltà nell'esecuzione di una puntura lombare.
 Pelle normale e ispessita nella SLA
Pelle normale e ispessita nella SLA Criteri di El Escorial rivisti per la SLA (1998)
SLA affidabileè impostato quando i segni di danno ai motoneuroni periferici e centrali sono combinati a tre livelli del sistema nervoso centrale su quattro possibili (tronco, cervicale, toracico e lombare).
Probabile- questa è una combinazione di segni a due livelli del sistema nervoso centrale. Alcuni segni di danno ai motoneuroni centrali sono molto elevati.
Probabile suscettibile di laboratorio- una combinazione di segni allo stesso livello del sistema nervoso centrale in presenza di segni di danno ai motoneuroni periferici in almeno due arti e assenza di segni di altre malattie.
Possibile SLA- una combinazione di caratteristiche allo stesso livello. O ci sono segni di danno al motoneurone centrale rostrale a segni di danno al motoneurone periferico, ma non ci sono dati ENMG ad altri livelli. Richiede l'esclusione di altre malattie.
Sospettare- questi sono segni isolati di danno al motoneurone periferico in due o più parti del sistema nervoso centrale.
Progressione della malattia del motoneurone
La progressione della malattia del motoneurone può essere suddivisa in tre tipi:
- Rapido: perdita di oltre 10 punti in 6 mesi
- Media - perdita di 5 - 10 punti per sei mesi
- Lento - perdita di meno di 5 punti in un mese
 Progressione della malattia del motoneurone
Progressione della malattia del motoneurone Metodi strumentali per la diagnosi della SLA
I metodi diagnostici strumentali sono progettati per escludere malattie potenzialmente curabili o con prognosi benigna.
 L'elettromiografia nella diagnosi della SLA
L'elettromiografia nella diagnosi della SLA Esistono due metodi di diagnostica ALS:
- (EMG) - verifica carattere generalizzato processi
- Risonanza magnetica(MRI) del cervello e del midollo spinale - esclusione delle lesioni focali del sistema nervoso centrale, le cui manifestazioni cliniche sono simili a quelle dell'insorgenza della MND
Sclerosi laterale amiotrofica - Trattamento
Sfortunatamente, al momento non esiste un trattamento completo per la sclerosi laterale amiotrofica. Pertanto, questa malattia è ancora considerata incurabile. Almeno, non sono ancora stati registrati casi di guarigione dalla SLA.

Ma la medicina non si ferma!
Vari studi sono costantemente in corso. Le cliniche utilizzano metodi che aiutano il paziente ad alleviare la tolleranza della malattia. Discuteremo queste linee guida cliniche di seguito.
Ci sono anche farmaci che prolungano la vita di un paziente con SLA.
Ma vale la pena dire che non è necessario effettuare il trattamento con metodi che non sono stati ancora studiati a fondo. Ci sono alcuni farmaci che a prima vista possono migliorare le condizioni del paziente. Tuttavia, nel tempo, tutto torna al punto di partenza e la malattia inizia ancora a progredire.
Lo stesso vale per cellule staminali. Sono stati condotti studi che hanno mostrato un miglioramento all'inizio. Tuttavia, poi, la persona è peggiorata e la malattia di SLA ha ricominciato a progredire.
Pertanto, al momento, le cellule staminali non sono la modalità di trattamento da scegliere. Inoltre, la procedura stessa è molto costosa.
Terapia patogenetica per la malattia di Lou Gehrig
Ci sono farmaci che rallentano la progressione della malattia di Lou Gehrig.
- inibitore presinaptico del rilascio di glutammato. Allunga la vita dei pazienti in media di 3 mesi. Devi prendere mentre una persona mantiene il self-service. Dosi di 50 mg 2 volte al giorno prima dei pasti ogni 12 ore.
 Riluzolo (Rilutek)
Riluzolo (Rilutek) Nel 3-12% dei casi, il farmaco provoca epatite farmaco-indotta, aumento della pressione. Metabolizzato negli uomini e nei fumatori. Hanno bisogno di una dose maggiore.
Vale la pena dire che il rallentamento della progressione non si fa sentire. Il farmaco non rende una persona migliore. Ma il paziente sarà malato più a lungo e in seguito smetterà di servirsi.
Il farmaco è controindicato nei pazienti con SLA certa e probabile con una durata della malattia inferiore a cinque anni con una capacità vitale forzata superiore al 60% e senza tracheostomia.
Il prezzo di Riluzole in diverse farmacie varia da 9.000 a 13.000 rubli.
NP001 — sostanza attiva clorito di sodio. Questo medicinale è un immunoregolatore per le malattie neurodegenerative. Sopprime l'infiammazione dei macrofagi in vitro e nei pazienti con sclerosi laterale amiotrofica.
 clorito di sodio
clorito di sodio 3 mesi dopo il contagocce alla dose di 2 mg/kg, il clorito di sodio stabilizza il decorso della malattia. Sembra fermare la progressione.
Cosa non può essere applicato
Per la SLA, non utilizzare:
- Citostatici (immunodeficienza aggravata nella malnutrizione)
- Ossigenazione iperbarica (aggrava il washout di anidride carbonica già disturbato)
- Infusioni saline fisiologiche per l'iponatriemia in pazienti con SLA e disfagia
- Ormoni steroidei (causano miopatia dei muscoli respiratori)
- Amminoacidi a catena ramificata (abbreviano la vita)
Terapia palliativa per la malattia di Charcot
L'obiettivo della terapia palliativa per la malattia di Charcot è ridurre i sintomi individuali. Oltre a prolungare la vita del paziente e mantenere la stabilità della sua qualità in un determinato stadio della malattia.
- Trattamento dei sintomi fatali (disfagia, insufficienza nutrizionale e respiratoria)
Trattamento dei sintomi non fatali della SLA
Consideriamo ora il trattamento della SLA con sintomi non fatali.

Per cominciare, questo riduzione di fascicolazioni e crampi:
- Chinidina solfato (25 mg due volte al giorno)
- Carbamazepina (100 mg due volte al giorno)
Può somministrare farmaci riducendo il tono muscolare:
- Baclofene (fino a 100 mg al giorno)
- Sirdalud (fino a 8 mg al giorno)
- Rilassanti muscolari azione centrale(diazepam)
Combattere le contratture articolari:
- Scarpe ortopediche (prevenzione della deformità del piede equinovaro)
- Compresse (novocaina + dimexide + idrocortisone / lidasi / ortofen) per il trattamento della periatrosi omeroscapolare
Farmaci metabolici miotropici:
- Corso di carnitina (2 - 3 g al giorno) 2 mesi, 2 - 3 volte l'anno
- Il corso di creatina (3 - 9 g al giorno) per due mesi passa 2 - 3 volte l'anno
Danno anche preparati multivitaminici. Neuromultivit, milgamma, preparazioni di acido lipoico per fleboclisi. Il corso del trattamento è di 2 mesi e 2 volte l'anno.
Trattamento della fatica:
- Midantan (100 mg al giorno)
- Etosuccimide (37,5 mg al giorno)
- Fisioterapia
Per la prevenzione della sublussazione della testa omero con paresi flaccida delle mani si utilizzano bende di scarico per gli arti superiori del tipo Deso. Devi indossare 3-5 ore al giorno.
Ci sono speciali ortesi. Questi sono supporti per la testa, supporti per arresto e stecche per le mani.

Esistono anche ausili sotto forma di stampelle, deambulatori o cinture per il sollevamento di un arto.
 Dispositivi ausiliari
Dispositivi ausiliari Ci sono anche utensili e dispositivi speciali che facilitano l'igiene e la vita di tutti i giorni.
 Dispositivi igienici per pazienti con SLA
Dispositivi igienici per pazienti con SLA Trattamento della disartria:
- Raccomandazioni sul discorso
- Applicazioni di ghiaccio
- Somministrare farmaci che riducono il tono muscolare
- Usa tabelle con alfabeto e dizionari
- Macchine da scrivere elettroniche
- Registratori vocali multifunzionali
- Amplificatori vocali
- Un sistema informatico con sensori accesi bulbi oculari per riprodurre il parlato come testo sul monitor (figura sotto)
 Terapia dei disturbi del linguaggio per le persone con malattia di Charcot
Terapia dei disturbi del linguaggio per le persone con malattia di Charcot Polmonite da aspirazione nella SLA
Il 50-75% dei pazienti con neurodegenerazioni e gli anziani hanno problemi di deglutizione, che si verificano nel 67% dei pazienti con SLA. Nel 50%, la polmonite da aspirazione è fatale.
Metodi diagnostici:
- Videofluoroscopia, scale APRS (scala di aspirazione-penetrazione) DOSS (scala di gravità dell'esito della disfagia)
Di seguito vediamo la definizione della densità e del volume di liquido, nettare e budino, che il paziente deve deglutire.
 Test di volume e densità dei poli
Test di volume e densità dei poli Trattamento della disfagia
Nel trattamento della disfagia (disturbo della deglutizione) su stato iniziale applicare quanto segue:
- Alimenti di consistenza semisolida con mixer, frullatore (purè di patate, gelatina, cereali, yogurt, gelatina)
- Addensanti fluidi (risorsa)
- Sono esclusi i piatti difficili da deglutire: con fase solida e liquida (zuppa con pezzi di carne), cibi solidi e sfusi (noci, patatine), cibi viscosi (latte condensato)
- Ridurre i prodotti che aumentano la salivazione (latte fermentato, caramelle dolci)
- Escludere i prodotti che causano un riflesso della tosse (condimenti piccanti, alcol forte)
- Aumentare il contenuto calorico del cibo (aggiungendo burro, maionese)
La gastrostomia è usata per trattare la disfagia progressiva. In particolare, endoscopica percutanea.
 Fasi della gastrostomia endoscopica percutanea
Fasi della gastrostomia endoscopica percutanea La gastrostomia percutanea endoscopica (PEG) e la nutrizione enterale prolungano la vita dei pazienti con SLA:
- Gruppo SLA + PEG — (38 ± 17 mesi)
- Gruppo SLA senza PEG — (30 ± 13 mesi)
Trattamento dell'insufficienza respiratoria
Il trattamento dell'insufficienza respiratoria è la ventilazione intermittente non invasiva (NIPPV, BIPAP, NIPPV), pressione positiva a due livelli (pressione inspiratoria superiore alla pressione espiratoria).
Indicazioni per la SLA:
- Spirografia (FVC)< 80%)
- Manometria inspiratoria - inferiore a 60 cm. Arte.
- Polisonnografia (più di 10 episodi di apnea all'ora)
- Pulsossimetria (Pa CO2 ≥ 45 mmHg; diminuzione della saturazione notturna ≥ 12% in 5 minuti)
- pH sangue arterioso meno di 7.35
 Dispositivo di respirazione per pazienti con SLA
Dispositivo di respirazione per pazienti con SLA Indicazioni per autorespiratore:
- SLA spinale con capacità vitale forzata (FVC) 80-60% - sonno 22 (S)
- BASSO bulbare FVC 80-60% - sonno 25 (ST)
- SLA spinale FVC 60-50% – sonno 25 (ST)
- FVC inferiore al 50% - tracheostomia (dispositivo VIVO 40, VIVO 50 - PCV, modalità PSV)
La ventilazione artificiale (invasiva) è sicura, impedisce l'aspirazione e prolunga la vita umana.
Tuttavia, contribuisce alla secrezione, al rischio di infezione, alle complicazioni della trachea. Oltre al rischio di sindrome del lock-in, dipendenza 24 ore su 24 e costi elevati.
Indicazioni per la ventilazione invasiva:
- Incapacità di adattarsi a NPVL o dura più di 16-18 ore al giorno
- In disturbi bulbari ad alto rischio di aspirazione
- Quando NVPL non fornisce un'adeguata ossigenazione
Criteri che limitano il trasferimento alla ventilazione meccanica nella SLA:
- Età
- Tasso di progressione della malattia
- Possibilità di comunicazione
- Demenza frontotemporale
- Le relazioni in famiglia
- Malattia mentale in un paziente
- Ebbrezza abituale
- Paura della morte
I pazienti con SLA sottoposti a ventilazione meccanica da più di 5 anni sviluppano la sindrome del lock-in (18,2%), uno stato di socialità minima (33,1%).
 Comunicatore per la comunicazione
Comunicatore per la comunicazione Psicoterapia per la sclerosi laterale amiotrofica
La psicoterapia è anche molto importante per la sclerosi laterale amiotrofica. È necessario sia per il paziente che per i suoi familiari.

Secondo le statistiche, l'85% dei pazienti con SLA soffre di disturbi mentali. E il 52% soffre i familiari. Allo stesso tempo, i parenti sono dominati da disturbi d'ansia e nei pazienti con depressione.
A disposizione segnali di allarme e da dipendenza. Cioè, l'abuso di tabacco, droghe o alcol. I pazienti rappresentano il 49% e i familiari l'80%. Di conseguenza, tutto ciò porta alle seguenti conseguenze.
Patogenesi dei disturbi mentali nella SLA
Quindi, cosa succede alla psiche umana sul palco diagnosi:
- Pensiero ambivalente (diviso).
- Sviluppo dell'ansia - dipendenza da droghe e Internet
- Pensieri ossessivi - sindrome ossessiva (ripetizione di esami con diversi medici)
- Violazione stato mentale parenti - disturbi del comfort (bozzolo psichiatrico)
Sul palco sviluppo del deficit neurologico:
- Negazione della malattia (reazione simile all'isteria invertita)
- Oppure c'è un'accettazione della malattia (depressione)
Sul palco deficit neurologico crescente:
- Depressione profonda
- Rifiuto del trattamento
Trattamento dei disturbi mentali
In generale, in questo caso, con la malattia di Charcot, è imperativo affrontare il trattamento dei disturbi mentali. Ciò comprende:
- Sostituzione di anticolinergici contro la salivazione (atropina, amitriptilina) con butolotossina e irradiazione delle ghiandole salivari
- colinomimetici (basse dosi di galantamina)
- Antipsicotici atipici - seroquel, possibilmente in gocce - neuleptil
- Non iniziare il trattamento con antidepressivi, poiché aumentano l'ansia. È meglio dare qualcosa di più morbido (azafen).
- Uso di tranquillanti - alprazolam, strezam, mezapam
- Sonniferi da usare solo se immobilizzati
- Pantogami
Terapia Occupazionale e Fisioterapia per la SLA
Parliamo un po' di terapia occupazionale e fisioterapia per la SLA. Scoprirai cos'è e a cosa serve.

Fisioterapista aiuta il paziente a mantenere una forma fisica e una mobilità ottimali, tenendo conto di come tutto ciò influisca sulla vita.
Se la terapia fisica controlla come cambia la vita del paziente, allora è necessaria un'altra persona che valuterà la qualità stessa della vita del paziente.
Ergoterapistaè uno specialista che aiuta il paziente a vivere nel modo più indipendente e interessante possibile. Lo specialista dovrebbe approfondire le peculiarità della vita del paziente. Soprattutto per i pazienti con casi gravi della malattia.
Smagliature
Stretching e movimenti di massima ampiezza prevengono le contratture, riducono la spasticità e il dolore. Comprese le contrazioni convulsive involontarie.
Puoi fare stretching con l'aiuto di un'altra persona o con una forza aggiuntiva esterna. Il paziente può farlo da solo con l'aiuto delle cinture. Ma qui devi ricordare che con questo metodo una persona sprecherà energia.
L'allenamento muscolare aiuta con la SLA?
Ci sono studi che affermano che l'uso di esercizi di resistenza per gli arti superiori in un paziente con sclerosi laterale amiotrofica, ha portato ad un aumento della forza statica in 14 gruppi muscolari (in 4 non sono aumentati).
E questo dopo 75 giorni di allenamento. Pertanto, qualsiasi lezione dovrebbe essere regolare e lunga.
Progressi nella forza muscolare mostrati nello studio
Come puoi vedere, c'è stato un aumento significativo della forza muscolare nella maggior parte dei gruppi. Come risultato di questa attività, è diventato più facile per una persona prendere gli articoli dallo scaffale più alto. Oppure prendi un oggetto e mettilo su uno scaffale.
Nella terapia stessa, i movimenti sono stati eseguiti lungo una traiettoria vicina a quella utilizzata nel PNF. Questo è il momento in cui il medico oppone resistenza al movimento del paziente. Naturalmente, tale resistenza non dovrebbe essere molto forte. La cosa principale è prendere il giusto grado.

Di norma, molti movimenti vengono eseguiti in diagonale. Ad esempio, prendi la gamba in diagonale. Gli esercizi stessi sono stati eseguiti con l'aiuto di elastici (tamburi). Sebbene siano stati in parte realizzati con l'aiuto della resistenza con le mani.
 Esercizi con elastici (batteria)
Esercizi con elastici (batteria) Metodologia per lo svolgimento delle lezioni
Ecco la metodologia di allenamento:
- L'azione è stata eseguita con il superamento della resistenza durante tutto il movimento
- Il movimento in una direzione ha richiesto 5 secondi
- La sessione consisteva in due serie di 10 movimenti.
- Il riposo tra le serie era di circa 5 minuti.
- Le lezioni si sono svolte 6 volte a settimana:
- 2 volte con uno specialista (superando la resistenza delle mani di uno specialista)
- 2 volte con un familiare (stessa resistenza del braccio)
- 2 volte da solo con bende elastiche
- I risultati della tabella sono apparsi dopo 75 giorni (65 lezioni) di corso
Qual è l'effetto della forza
Perché la forza aumenta anche quando un motoneurone è danneggiato? Uno stile di vita sedentario può portare a detraining cardiovascolare.
In altre parole, anche quei muscoli che non sono colpiti iniziano a indebolirsi. Ed è sul loro allenamento che si basa l'effetto dell'aumento della forza.
Un altro detraining si verifica perché cade supporto funzionale. In modo sedentario vita, c'è poco ossigeno. Pertanto, esercizi con moderato attività fisica migliorare il funzionamento generale del corpo.
Intensità dell'allenamento nella malattia di Lou Gehrig
Voglio dire che gli esercizi con forte resistenza non danno alcun effetto. Non solo non danno, ma possono anche danneggiare un paziente con la malattia di Lou Gehrig.
Ma esercizi di moderata intensità portano a un miglioramento del lavoro dei muscoli che non sono affetti da debolezza significativa. Se il muscolo praticamente non si muove, non dovresti contare su un aumento della sua forza.
Tuttavia, ci sono muscoli che fanno movimenti, ma debolmente.
Anche le persone con insufficienza respiratoria coloro che utilizzano la ventilazione assistita non invasiva possono avere miglioramenti nella funzione muscolare come risultato di un esercizio di intensità moderata.
L'esercizio fisico regolare con resistenza moderata aiuta a migliorare la forza statica in alcuni muscoli.
Per determinare il carico significativo e insignificante, viene utilizzata la scala Borg.
Scala Borg
Da 0 a 10, possiamo determinare le sensazioni soggettive che una persona ha quando fa esercizi.
Non importa quanto sforzo facciano i pazienti. Non importa quali esercizi e la durata della passeggiata sono stati offerti. Soprattutto, il loro sforzo dovrebbe sembrare nient'altro che "medio" o "quasi pesante". Cioè, il carico non dovrebbe superare il livello 4.
Esiste un tale calcolo che aiuta a determinare la riserva del cuore. Cioè, a quale frequenza cardiaca puoi allenarti facilmente e in sicurezza.
- Intensità di carico= Frequenza cardiaca a riposo + dal 50% al 70% di riserva cardiaca
- Riserva del cuore= 220 - età - frequenza cardiaca a riposo
Programma di allenamento aerobico per la SLA
C'è anche un programma di allenamento aerobico di 16 settimane per la SLA. Si fa tre volte a settimana. Un paio di volte lo facciamo a casa su un ergometro da bicicletta e nella steppa. Poi una volta in ospedale sotto la supervisione di un fisioterapista.

- 1-4 settimane da 15 a 30 minuti
- Da 5 settimane a mezz'ora
Esercizi per passi(un gradino che si sale e poi si scende):
- Prime 5 settimane (3 minuti)
- 6-10 settimane (4 minuti)
- 11-16 settimane (5 minuti)
Classi in ospedale:
- 5 minuti di riscaldamento (pedalare senza carico)
- 30 minuti di esercizio moderato (15 minuti in bicicletta, 10 minuti su pista, 5 minuti di passo)
- 20 minuti di allenamento di forza (anteriore coscia, bicipiti e tricipiti)
- 5 minuti della parte finale
E questo programma ha portato anche al fatto che i pazienti hanno poi migliorato le loro condizioni fisiche.
Principi per il ripristino delle attività quotidiane
L'esercizio fisico e il terapista occupazionale dovrebbero sempre pensare a tre cose per migliorare le attività quotidiane di una persona:
- Lo specialista deve valutare e ottimizzare le capacità fisiche del paziente
- Date queste possibilità, è necessario selezionare la postura ottimale per ogni azione.
- Adattare l'ambiente del paziente e vedere come le capacità fisiche possono essere migliorate
Supporto per la deambulazione per la malattia di Charcot
Molto spesso, gli specialisti devono risolvere il problema di sostenere la deambulazione del loro paziente con malattia di Charcot. Ci sono diverse soluzioni qui. Puoi usare le stampelle per i gomiti perché sono più facili da appoggiare.

Ma a volte è sufficiente usare le normali canne. Ma richiedono forza perché devi tenere tutta la mano. E per camminare su superfici scivolose, consiglio di acquistare in farmacia speciali "gatti".
 Ramponi per camminare su superfici scivolose
Ramponi per camminare su superfici scivolose Se il piede si piega, è necessario un supporto per l'arresto.
Se una persona è troppo stanca o semplicemente non può camminare, sarà necessaria una sedia a rotelle. E qual è il modello ottimale da scegliere: questo problema dovrebbe essere deciso congiuntamente dall'ergoterapista e dal fisioterapista.

In una sedia a rotelle, una persona dovrebbe essere comoda, comoda. Inoltre, il dispositivo stesso non dovrebbe richiedere molta energia dal paziente.
È inoltre necessario tenere conto della modularità (opzioni) del passeggino. Aiutano a svolgere le attività quotidiane.
Ad esempio, una sedia a rotelle può utilizzare un dispositivo antiribaltamento per impedirne la caduta. Possono essere utilizzate anche ruote di trasporto. Quindi puoi guidare dove un passeggino largo non si adatta.
A volte dobbiamo fornire freni per il manutentore. Possono essere inclusi anche poggiatesta, cinture laterali e così via.
Come puoi vedere, anche l'adattamento e la scelta di una sedia a rotelle è una parte molto importante per un paziente con SLA.
altezza del sedile
Anche l'altezza del sedile è importante. Da una superficie più alta sarà molto più facile per il paziente alzarsi. Anche ad alto livello, una persona siederà in modo più uniforme. Infatti, in questo caso, vengono attivati i muscoli della schiena.
Pertanto, non è necessario far sedere il paziente su una sedia bassa. Si sentirà a disagio. Inoltre, sarà molto più difficile rimuovere un paziente da una sedia del genere.
 Regolazione dell'altezza del letto
Regolazione dell'altezza del letto Quindi, oltre al passeggino, devi regolare il livello ottimale del letto e della sedia.
pasto
Mangiare dipende dal tavolo a cui è seduto il paziente. Evita i tavoli rotondi perché rendono difficile la seduta.
Se una persona ha una testa cattiva e il cibo gli cade dalla bocca, in nessun caso fare una testiera alta. Questo gli renderà molto difficile deglutire.

Se metti il paziente su una testiera alta, assicurati almeno che tutta la sua schiena si trovi su questa testiera. Cioè, la schiena non dovrebbe piegarsi.
Se una persona giace completamente orizzontale, avrà una capacità respiratoria molto bassa. Se viene alzato di 30º, sarà molto meglio.
Nella posizione laterale e su testiera alta sarà meglio che solo di fianco. Ma la posizione peggiore è sulla schiena e in orizzontale.
Se sei seduto, la respirazione sarà ancora migliore! Ricorda che mangiamo sempre seduti. Pertanto, inclinare la testa in avanti aiuta sempre la faringe.
Ci sono anche molte posate adattate che sono più facili da tenere in mano. Ad esempio, dispositivi con manici ispessiti. Ci sono anche morsetti speciali sulla spazzola e molto altro.
Ma fai attenzione se la persona ha disfagia! In questo caso, devi decidere se mantenere attive le prestazioni amatoriali o un sorso sicuro. Quest'ultimo avrà sempre la precedenza.
Se una persona ha la disfagia, sarà difficile per lui pensare a una gola fittizia e, inoltre, portare questo cibo a se stesso con una mano debole. Pertanto, in questo caso, non è necessario inseguire l'adattamento al self-service.
Bere
Ci sono posate che aiutano a svolgere il bere. Ad esempio, ci sono occhiali speciali con un taglio. Di conseguenza, puoi bere da un bicchiere del genere senza gettare indietro la testa. Il naso scende in questa tacca, rendendo il bere più sicuro.
 Occhiali ritagliati
Occhiali ritagliati C'è anche una tazza con due manici. È molto comodo da trasportare con due mani.
 Tazza con due manici
Tazza con due manici Tuttavia, un capezzolo speciale sul coperchio superiore può anche essere pericoloso per la disfagia. Se bevi da un bicchiere del genere, una persona dovrà gettare indietro la testa.
Programma di posizionamento ALS
Per coordinare il regime di lavoro e di riposo, nonché per bilanciare le capacità del malato di SLA dall'eccessivo affaticamento, è necessario attenersi a programma speciale(orari).
Qui programma di esempio posizionamento per una persona gravemente menomata che potrebbe rimanere in posizione eretta per qualche tempo.
Malattia SLA - foto
Di seguito una foto relativa alla malattia di SLA. Tutte le immagini sono cliccabili per ingrandire.
















